Hemostasis and Coagulation
Hemostasis, the stoppage of bleeding, may involve three basic reactions: (1) constriction by the smooth muscle of the injured vessel to reduce the opening, (2) formation of a platelet plug to occlude the opening, and (3) clot formation to complete occlusion of the opening.
injuries to vessels do not require the formation of a clot (coagulation) if hemostasis can be achieved by the first two reactions.Platelets and the Endothelium
When a vessel is injured and the endothelial cell lining is damaged so that the underlying connective tissue is exposed, platelets adhere to collagen and other proteins in the connective tissue. This platelet adhesion results from binding between platelet cell membrane proteins and the connective tissue. The cell membrane of adhered platelets undergoes alterations, and secretory granules are also released. Platelets that have undergone these reactions are termed activated platelets. The presence of activated platelets stimulates other platelets to adhere to those already present. The collection of platelets forms a platelet plug that may be sufficient (together with local vasoconstriction) to occlude extremely small openings in damaged vessels and bring about hemostasis. Platelet aggregation is the term applied to the overall sequence of events responsible for the formation of the platelet plug.
Platelet aggregation is also subject to regulation by two different eicosanoids, thromboxane A2 (TXA2) and prostacyclin (PGI2). TXA2 is a stimulant of platelet aggregation, and prostacyclin inhibits platelet aggregation. The primary source of TXA2 in the area of a damaged vessel is adhered platelets that increase their synthesis of TXA2 after adhesion. The primary source of prostacyclin is intact, undamaged endothelial cells. As the platelet plug grows and extends to areas where endothelial cells remain intact, the local concentration of prostacyclin generated by undamaged endothelial cells acts to stop the growth of the platelet plug.
TXA2 and serotonin (also released by adhered platelets) are both vasoconstrictors, stimulating smooth muscle contraction to assist with hemostasis.Aspirin inhibits the formation of eicosanoids by binding to and inhibiting cyclooxygenase, an enzyme necessary for their synthesis. TXA2 and prostacyclin are both eicosanoids, so aspirin initially reduces the formation of both. However, platelets do not have nuclei and cannot synthesize new enzymes, while nucleated endothelial cells can synthesize additional cyclooxygenase. Because of these differences in the two cell types, treatment with aspirin at an appropriate dose and an appropriate schedule preferentially reduces thromboxane synthesis by platelets. This is the basis for the use of aspirin when it is desirable to reduce the tendency for blood coagulation (e.g., humans with cardiac diseases and animals with heartworms.)
some of the substances released by aggregating platelets and some of the cell membrane components exposed by aggregated platelets act to promote coagulation. Thus, with larger sites of injury, more platelets aggregate and more stimulants of clotting are present in the local area. Coagulation is initiated when the local concentration of these substances reaches some critical level. Platelets are required for normal coagulation in response to vascular damage. Clotting may also be stimulated outside of the body when a foreign surface, such as the glass surface of a test tube, induces the same reactions as exposure to collagen.
Intrinsic and Extrinsic Coagulation Pathways
The ultimate product of blood coagulation is a relatively solid gel plug (clot or thrombus). This plug may be red or somewhat clear. The color varies with the number of erythrocytes and other blood cells trapped in the clot. Erythrocytes and leukocytes are not necessary for coagulation, and they may or may not be present in a clot.
A clot is relatively solid because of interlacing strands of fibrin (a protein polymer) that are covalently cross-linked.
Trapped within this network are other blood components (e.g., platelets, leukocytes, erythrocytes), but the key element is the fibrin. Thus, the most basic explanation of coagulation is that it is a series of biochemical reactions to produce and stabilize a protein polymer, fibrin.The series or chain of biochemical reactions that links initial exposure to collagen or a surface other than normal endothelium (e.g., glass surface of a blood draw tube) to a stabilized fibrin network is the intrinsic clotting pathway, or intrinsic cascade (Fig. 15-4). It is intrinsic because all substances necessary for the cascade are present in the circulation. This pathway includes several proteolytic enzymes (clotting factors) normally in the plasma in an inactive form. When one of these inactive forms converts to an active form, it activates the next
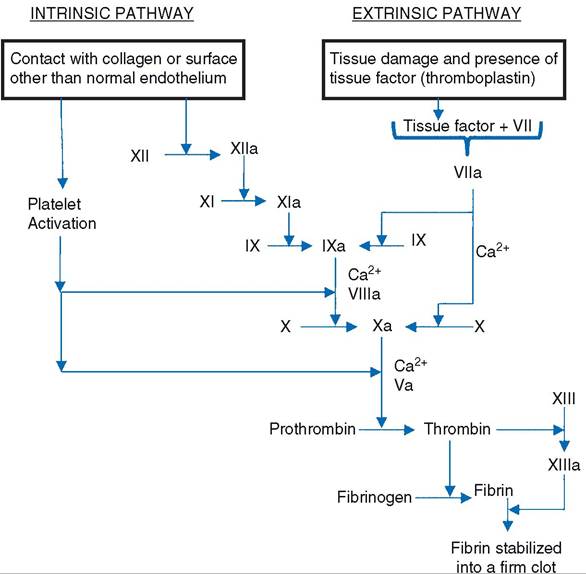
Figure 15-4. Intrinsic and extrinsic pathways leading to the generation and stabilization of fibrin, which forms the framework of a clot. Two factors, VIII and V, are cofactors and are shown only in their active forms. Note that Ca2+ and activated platelets are cofactors at several points in the cascade.
Table 15-3. International Nomenclature of Coagulation Factors with Synonyms
Factora Synonyms
I Fibrinogen
II Prothrombin
III Tissue thromboplastin
IV Calcium
V Proaccelerin; labile factor
VII Proconvertin; stable factor
VIII Antihemophilic globulin; antihemophilic factor A
IX Christmas factor; antihemophilic factor B
X Stuart-Power factor
XI Plasma thromboplastin antecedent; antihemophilic factor C
XII Hageman factor
XIII Fibrin stabilizing factor aInternational nomenclature.
enzyme in the cascade. This type of cascade allows for amplification at multiple steps so that many molecules of fibrin can be generated as the result of the activation of one initial molecule.
The factor numbers refer to the sequence in which they were discovered, not the sequence of their involvement in the cascade. The numbered factors have also been identified by common name (Table 15-3). As shown in Figure 15-4, the first step in the intrinsic cascade is the activation of factor Xii. This could occur when a vessel is damaged and the underlying tissue is exposed or when blood is drawn into an untreated glass tube.Coagulation can also be initiated when a protein from interstitial fluid (tissue factor or tissue thromboplastin) forms an active complex with an inactive plasma protein, factor VII (Fig. 15-4). Tissue thromboplastin is a component of cell membranes of various cell types and apparently may be released from injured cells. The factor VII-tissue factor complex activates factors X and IX. Factor X is a component of the intrinsic cascade, so from this point on, the pathway to fibrin formation and linkage is the same as in the intrinsic cascade. The cascade that is initiated by tissue thromboplastin is the extrinsic cascade, or extrinsic pathway. The final product of both the extrinsic and intrinsic cascades is a fibrin clot, and some clotting factors are common to both. The only differences are some of the factors found in the early steps of the cascades (Fig. 15-4).
Calcium ions are required as cofactors at various steps in both cascades (Fig. 15-4), and several anticoagulants used to prevent blood clotting outside the body do so by binding calcium ions. These include sodium citrate, potassium citrate, ammonium citrate, and ethylenediaminetetraacetic acid (EDTA). EDTA is usually in the form of a sodium or potassium salt.
Clot formation at the site of injury reduces blood loss and occludes the opening in the damaged vessel. This tends to remove stimuli necessary for continuation of coagulation by covering the exposed collagen and preventing the entry of tissue fluid into the vessel. The undamaged endothelial cells adjacent to the damaged area also secrete prostacyclin, an inhibitor of platelet adhesion and aggregation.
If coagulation continued unchecked beyond the site of injury, blood vessels throughout the body would be occluded inappropriately, and blood flow would be halted. Two additional plasma proteins, protein C and antithrombin III, prevent coagulation from continuing inappropriately. Protein C circulates in an inactive form that is activated by thrombin. Thrombin forms as part of both the intrinsic and extrinsic pathways and is also responsible for one of the final steps in both, the production of fibrin from fibrinogen (Fig. 15-4). Thus, as formation of thrombin by the coagulation pathways promotes clot formation, thrombin also restrains the process so that it does not become uncontrolled. Antithrombin III is also inactive by itself, but when bound to heparin (present on normal endothelial cell membranes), the heparin-antithrombin III combination inactivates thrombin. Here again, the presence of intact, healthy endothelial cells acts to prevent or halt coagulation. Inhibition or inactivation of thrombin is a very efficient means of inhibiting coagulation in that thrombin has a positive feedback effect on coagulation by activating several clotting factors that precede it in the cascade.In many cases, the damage to small vessels can be repaired so that normal blood flow can again occur. These repair mechanisms include removal of the clot and proliferation of endothelial cells to reestablish normal vascular lining. A key element in clot removal is the activation of the fibrinolytic system. This system is similar to the clotting cascade in that an inactive plasma protein or proenzyme, plasminogen, is activated to plasmin, which converts fibrin to soluble fragments. There appear to be several different pathways by which plasminogen can be activated to plasmin, but these are not as well understood as the activation of clotting factors. However, the presence of fibrin may stimulate several of these pathways. It seems that as fibrin is generated, it also begins to initiate its own ultimate destruction.
Plasminogen is also activated by tissue plasminogen activator, an enzyme secreted by normal, intact endothelial cells.The majority of the plasma factors for both the coagulation and fibrinolytic cascades are synthesized in the liver, and this synthesis is vitamin K dependent. Vitamin K is a lipid- soluble essential vitamin whose normal intestinal absorption requires bile salts. If the liver is diseased or the bile duct is obstructed, the resultant lack of bile in the gut reduces the absorption of lipids and vitamin K. Bacteria in the mammalian intestine also synthesize absorbable vitamin K.
Dicumarol is a toxic form of a general class of compounds known as coumarins. Dicumarol, found in spoiled sweet clover hay or silage, inhibits the clotting of blood. It is antagonistic to vitamin K and reduces the synthesis of vitamin K-dependent clotting factors. Among these is prothrombin. Sweet clover poisoning is the hemorrhagic condition resulting from excessive dicuma- rol ingestion. Clinical signs are related to reductions in the ability of blood to clot. Small cuts or bruises may result in bleeding that is difficult to stop. Warfarin is another coumarin and vitamin K antagonist that is used commercially in rodent poisons.