HEMOSTASIS: PREVENTION OF BLOOD LOSS
1. What is the sequence of events from the time of vascular injury to a return to normal?
2. What is the principal chemical component of the clotting factors? Note the major sites of their synthesis.
3. What vitamin is required for the synthesis of several coagulation factors?
4. What chemical element is required for nearly all the hemostatic reactions?
5. What is the substance contained in the basement membrane of capillaries and throughout the interstitial space that provides for platelet adhesion?
6. What are the properties of vascular endothelium that prevent activation of platelets and procoagulants?
7. What is another name for platelets?
8. Study the fine structure of the platelet and relate its structure.to the release of the granular contents.
9. What is the first response of platelets to disrupted endothelium and contact with subendothelial tissues?
0. In addition to collagen, what substance is required for the initial adhesion of platelets?
11. What is the principal messenger that is formed after platelet stimulation that will release Ca2+ from granule storage?
2. What is the role of aspirin in the blood coagulation scheme?
3. What is the platelet release reaction and how is it initiated?
4. What is accomplished by platelet aggregation?
15. What are the four key reactions involved in the formation of a clot?
6. What is the relationship of the tenase and prothrombinase.complexes to the formation of thrombin?
1.7. Know the difference between the extrinsic and intrinsic systems (tissue factor and contact activation pathways, respectively) and their relationship to each other.
8. In what way is the activation of factor X a focal point in the blood coagulation scheme?
9. What is the significance of factor XIII? What is its origin?
:o. How is clot retraction accomplished? What is its function?
’1.
Once initiated, what prevents blood coagulation from spreading (clot growth)?!2. What is the role of plasmin? What is the principal plasminogen activator?
The effectiveness of blood function depends on its circulation within a closed system of vessels. The vessels might open because of disease or accident, and blood loss can be prevented or minimized by hemostasis. Hemostasis is a complicated process and the following summary is provided as an orientation to the details that follow.
When a blood vessel is damaged, endothelial cells are separated, the underlying collagen is exposed, and the surface loses its usual smoothness and nonwettability. Often the vessel is torn, cut, or separated and the hemostatic crisis is exacerbated. Regardless of the severity, platelets begin to contact the damaged surface. This initiates the adhesion process because the platelets develop projections and become sticky. The adhered platelets undergo a reaction in which aggregating agents are released and cause the accumulation of more platelets. When this occurs, blood coagulation soon becomes evident at the damaged site, and the platelet plug is strengthened by the formation of a fibrin meshwork. Clot retraction (reduction in size) occurs and fibrinolysis
(dissolution of fibrin) begins. Finally, the damaged vessel is repaired by connective tissue and endothelial cell growth, and there is a return to normal when the platelet-fibrin complex and other cell debris is removed (Figure 3-11).
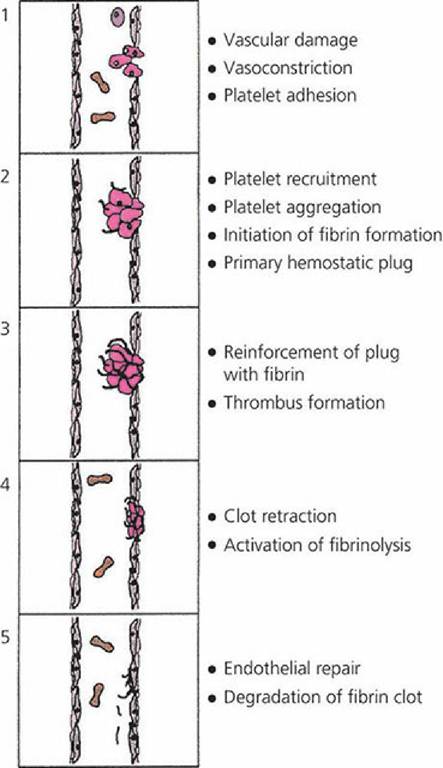
■ FIGURE 3-11 The five major stages in the formation and dissolution of a blood clot, or thrombus, around the site of vascular injury extending from the initiation of platelet activation after vascular damage through endothelial repair. (Adapted from Gentry PA. Blood coagulation and hemostasis. In: Reece WO, ed. Dukes’ Physiology of Domestic Animals. 12th edn. Ithaca, NY: Cornell University Press, 2004.
Used by permission of the publisher, Cornell University Press.)Hemostatic Components
A complex series of biochemical reactions make up the biochemical process. The major contributors to the process are proteins, vascular endothelium, and platelets.
Proteins
The protein components of the blood coagulation pathway, their synonyms or common abbreviations, are presented in Table 3-3. The designation of each factor with a roman numeral was described on first discovery and included factors I to XIII. As shown in Figure 3-3, many still persist, but the identification was discontinued (e.g., VI was initially described but found later not to exist; IV identified Ca2+ and was discontinued because it was not a protein). The major components now shown are proteins and the list has grown because of continued discovery. These proteins are present in the blood or tissues and simply await an activation mechanism.
TABLE 3-3 THE MAJOR COMPONENTS OF THE COAGULATION PATHWAY (ENZYMES, PROTEIN COFACTORS, AND SUBSTRATES) INVOLVED IN FIBRIN FORMATION AND FIBRIN DEGRADATION
| component | synonym | site of synthesis |
| Fibrinogen | Factor I | Liver |
| Prothrombin | bgcolor=white>Factor IILivera | |
| Thrombin | Plasma | |
| Tissue factor | Thromboplastin | Vascular endothelium |
| Factor V | Vascular endothelium | |
| Factor VII | Livera | |
| Factor VIII | Antihemophilic factor | Vascular endothelium |
| Factor IX | Christmas factor | Livera |
| Factor X | Stuart factor | Livera |
| Factor XI | Plasma thromboplastin antecedent | Liver |
| Factor XII | Hageman factor | Liver |
| Factor XIII | Fibrin stabilizing factor | Liver |
| von Willebrand factor | vWF | Vascular endothelium |
| Prekallikrein | PK, Fletcher factor | Liver |
| High-molecular-weight kininogen | HK, HMWK | Liver |
| Protein C | Livera | |
| Protein S | Livera | |
| Thrombomodulin | TM | Vascular endothelium |
| Plasminogen | Liver | |
| Tissue-type plasminogen activator | t-PA | Liver |
| Urokinase-type plasminogen activator | uPA, prourokinase | Unknown |
| aVitamin K-dependent protein. From Gentry PA. Blood coagulation and hemostasis. In: Reece WO, ed. Dukes’ Physiology of Domestic Animals. 12th edn. Ithaca, NY: Cornell University Press, 2004. Used by permission of the publisher, Cornell University. | ||
It is important to recognize that Ca2+ is required for nearly all the reactions and that vitamin K is required for production of prothrombin, protein C, protein S, and factors VII, IX, and X by the liver.
Vascular Endothelium
The entire cardiovascular system is lined by a single layer of flattened cells known as the endothelium. It not only lines the heart, but also the vessels. At the capillary level, all that remains is the endothelial layer. Regardless of its location, it is underlain with a basement membrane that contains collagen. Collagen fibers are also present throughout the interstitial space. Collagen in the subendothelial tissue, as well as fibronectin released from endothelial cells, provides for the adhesion of platelets to the site of vascular injury.
As long as the endothelium is intact, the platelets and the proteins associated with blood coagulation (procoagulants) are not activated. The properties of the endothelium that prevent activation include (1) the negative charge on the endothelial cell surface that repels the negatively charged platelets; (2) synthesis of inhibitors of platelet function (e.g., prostacyclin) and of fibrin formation (e.g., thrombomodulin); and (3) the generation of activators of,fibrin degradation (e.g., tissue plasminogen activator).
Platelets
Platelets are also known as thrombocytes. An appreciation of the complexity of the platelet can be obtained from Figure 3-13. The band of microtubules that encircles the platelet contracts when platelets are activated and results in change of shape and extrusion of platelet granule contents into the open canalicular system and subsequent release from the platelet to its exterior. The granules (alpha granules and dense granules) contain many of the coagulation factors, other proteins, calcium, serotonin, adenosine diphosphate (ADP), and adenosine triphosphate (ATP), all of which assist or potentiate the coagulation process.
Release of the granule contents requires energy from the mitochondria and glycogen particles and ionized calcium from the dense tubular system, a component of the membrane system of the platelet.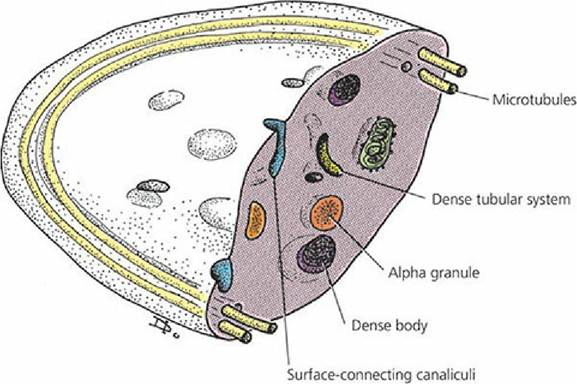
■ FIGURE 3-12 Internal details of a platelet discernible at the electron microscope level. Dense bodies are also known as dense granules. (From Cormack DH. Essential Histology. 2nd edn. Baltimore, MD: Lippincott Williams & Wilkins, 2001.)

■ FIGURE 3-13 Platelet adhesion. This is the first response to blood vessel injury. The platelets lose their discoid shape and form sticky projections (pseudopods) for their continued adherence to the injured vessel and entrapment of other platelets.
Platelet Reactions
Circulating platelets are recruited at the site of vascular injury, where they undergo structural changes. These changes are associated with platelet reactions and, coupled with the release of granule components, provide for a highly reactive surface for the formation of thrombin and fibrin.
PlateletAdhesion
The first response of platelets to disrupted endothelium and contact with subendothelial tissues is their adhesion or attachment to these surfaces. When this happens, a monolayer of platelets adheres to the site and they lose their discoid shape and form pseudopods, as shown in Figure 3-13. The pseudopods permit greater contact with other platelets flowing by the site of damage and also with those already adhering to the disrupted endothelium and exposed subendothelium. The initial adhesion requires collagen that is present in the subendothelium and fibronectin from the endothelial cells. Continued adhesion results from the von Willebrand factor (vWF) and fibronectin presence in platelet granules that are extruded from activated platelets.
PlateletActivation
This is the means whereby platelets are stimulated to begin their further role in assisting hemostasis.
The interaction of an agonist (e.g., collagen, thrombin, ADP) with its specific receptor on the platelet surface initiates the transmission of a signal through the cell membrane, which in turn activates intracellular messengers. Intracellular messenger activation results in the release of Ca2+ from storage pools into the platelet cytoplasm. The principal messenger, thromboxane A2 (TXA2), is produced from platelet membrane phospholipids after agonist interaction with the membrane receptors. Aspirin blocks the formation of TXA2, thus preventing the messenger from mobilizing Ca2 from the granules to the cytoplasm.Platelet Release Reaction
This event is initiated by the increase in intracellular calcium in response to the intracellular messenger and granular contents are secreted. It involves clustering of granular content into the center of the platelet after microtubular contraction and, finally, granule content extrusion to the exterior from the open canalicular system. The mechanisms of release are illustrated in Figure 3-14.
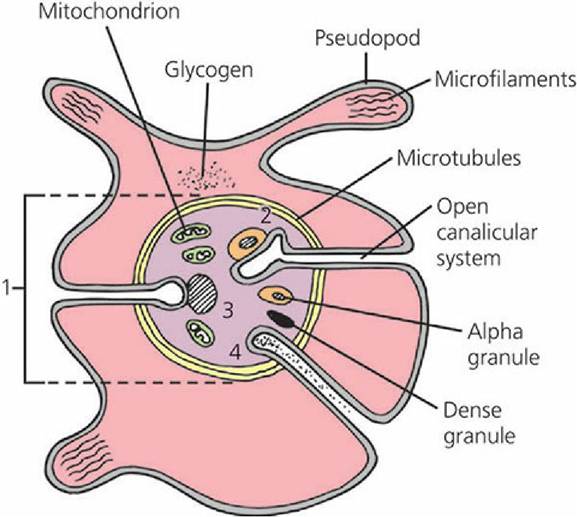
■ FIGURE 3-14 Platelet cross-section showing how microtubular contraction results in extrusion of platelet granule contents into the open canalicular system and release from the platelet. (1) Clustering of granules into the center of the platelet after microtubular contraction; (2) contact of granule membrane with open canalicular system membrane; (3) fusion of granule membrane with open canalicular system membrane; (4) granule content extruded from open canalicular system. (Modified from MacIntyre DE. The platelet release reaction: association with adhesion and aggregation and comparison with secretory responses in other cells. In: Gordon JL, ed. Platelets in Biology and Pathology, Vol. 1. Amsterdam: Elsevier, 1976.)
PlateletAggregation
The exterior presence of the granule contents provides high concentrations of fibrinogen (needed to form fibrin), fibronectin and vWF (both needed for adhesion), factor V, and other proteins that assist conversion of prothrombin to thrombin at the platelet surface, which, by piling platelets on each other, can lead to the formation of the primary platelet plug. After the release reaction, the platelets lose their individual integrity, lipoprotein membranes are fused, receptors are exposed for coagulation proteins (factors), and thus a highly reactive surface (platelet aggregates) is exposed for the formation of thrombin and fibrin.
Clot Formation (Blood Coagulation)2

Thrombin formation is the penultimate (next to last) stage in the formation of fibrin, which is insoluble and stabilizes the platelet plug. The stabilized platelet plug, formed by blood coagulation, is known as the secondary hemostatic plug or clot. Once the clot is formed, blood loss through the damaged endothelium is completely stopped. It was recognized previously that, after the platelet reactions, the stage was set for blood coagulation. Most of the proteins that participate in the hemostatic process circulate in plasma as inactive proenzymes, and each undergoes activation in sequence as coagulation proceeds. The sequence is referred to as a cascade phenomenon where each reaction represents an amplification point, whereby a small stimulus results in a larger response.
There are four key reactions involved in the formation of a clot: (1) activation of factor IX, (2) activation of factor X, (3) formation of thrombin, and (4) fibrin formation (Figure 3-15). Activated factor IX (FIXa) is a component of the tenase complex and activated factor X (FXa) is a component of the prothrombinase complex. These are key enzyme complexes assembled in close proximity on the surface of platelet aggregates. They accelerate the rate of biochemical cascade reactions resulting in the generation of thrombin (Figure 3-15).
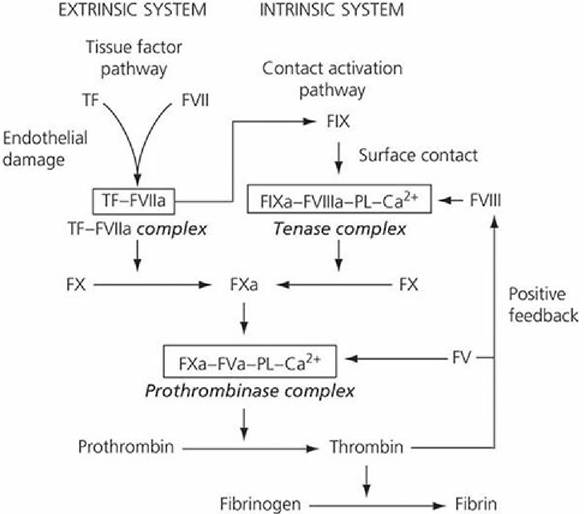
■ FIGURE 3-15 The two pathways by which factor X activation can occur. In the extrinsic pathway (tissue factor pathway), activated factor X (FXa) is generated by the direct action of the tissue factor (TF)-factor VIIa complex, whereas in the intrinsic pathway (contact activation pathway), factor IXa must combine with factor VIII, phospholipids (PL), and calcium to form the tenase complex before factor X can be activated at a physiologically relevant rate. The final common steps in fibrin formation involve the formation of the prothrombinase complex, which activates prothrombin, allowing thrombin to convert fibrinogen to fibrin. (From Gentry PA. Blood coagulation and hemostasis. In: Reece WO, ed. Dukes’ Physiology of Domestic Animals. 12th edn. Ithaca, NY: Cornell University Press, 2004. Used by permission of the publisher, Cornell University Press.)
V. Go to www.wiley.com/go/reece/functional to view a related video.
Pathways to Thrombin Formation
The conversion of prothrombin to thrombin (the key enzyme in hemostasis) is catalyzed by the prothrombinase complex (FXa, activated factors V [FVa], phospholipids [PL], and Ca2+). There are two separate activation mechanisms leading to the formation of the prothrombinase complex (see Figure 3-15); the tissue factor pathway (extrinsic system) and the contact activation pathway (intrinsic system). The tissue factor pathway begins with a traumatized vascular wall or traumatized extravascular tissues that come in contact with the blood. The contact activation pathway begins with trauma to the blood itself or exposure of the blood to collagen from a traumatized blood vessel wall. The pathways are not independent of each other and after blood vessel rupture, clotting occurs by both pathways simultaneously. Tissue factor (TF), also known as thromboplastin, initiates the tissue factor pathway (see Figure 3-15), whereas contact of factor XII and platelets with collagen in the vascular wall initiate the contact activation pathway (Figure 3-16).
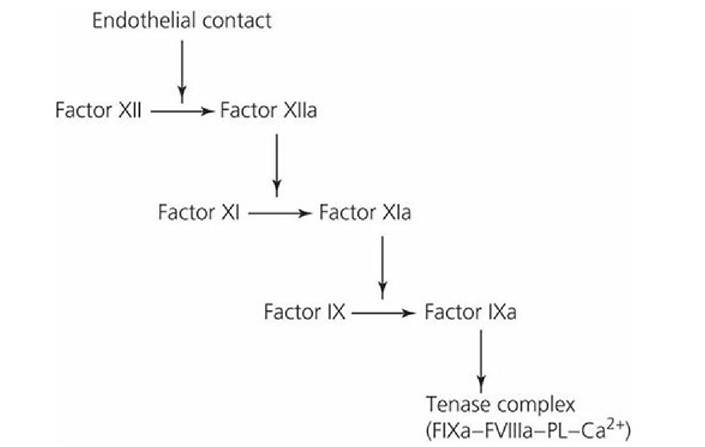
■ FIGURE 3-16 The contact phase for the activation of factor IX, initiated when factor XII is activated by contact with damaged endothelium. Factor XIIa activates factor XI (accelerated by prekallikrein [PK] and high-molecular-weight kininogen [HK]). Activated factor XI, in the presence of Ca2+, activates factor IX (FIXa). FIXa, in association with other components of the tenase complex, allows for the activation of factor X and FXa’s association with the prothrombinase complex. PL, phospholipid.
Following vascular damage, TF and binding sites for FVII, FIX, and FX are exposed on the surface of endothelial cells. In the presence of Ca2+, the TF-VIIa complex forms first and then activates FIX and FX (see Figure Figure 3-15). Activated FIX (FIXa) can then become a part of the tenase complex without having FIX being activated via factor XII in the contact activation pathway as shown in Figure 3-16. The rate of FXa formation by the proteolytic action of the tenase complex occurs at a much faster rate than that produced by the TF-VIIa complex acting alone and, accordingly, provides an amplification step in thrombin generation. In addition, the initial formation of thrombin accelerates FXa production by a positive feedback response that activates factor VIII, a component of the tenase complex, and factor V, a component of the prothrombinase complex (see Figure Figure 3-15). The contact activation pathway is required to sustain thrombin formation at the site of severe trauma.
After activation of FX, there is a common pathway to the formation of thrombin, after which fibrin is formed from fibrinogen (see Figure 3-15).
Fibrin Formation
The final step of blood coagulation is the conversion of fibrinogen (a plasma protein) to fibrin. This begins when thrombin has been formed. The first reaction produces fibrin monomers that spontaneously polymerize and a loosely knit mesh is formed, held together by covalent peptide bonds. This polymer structure is permeable to blood flow and is referred to as soluble fibrin. The
stabilization (formation of isopeptide bonds) of soluble fibrin to an insoluble fibrin clot is catalyzed by activated factor XIII (FXIIIa). Factor XIII is released from entrapped platelets and its conversion to the active form is induced by thrombin in the presence of calcium. Stabilization renders fibrin more elastic and less subject to lysis.
Clot Retraction
After stabilization, clot retraction (shrinking of the clot) occurs and is provided by the action of the platelet contractile proteins, thrombosthenin, actin, and myosin. These proteins are exposed when platelets are activated. The activation brings changes that activate thrombosthenin, actin, and myosin to react in a manner analogous to that which occurs during muscle contraction, and the clot retracts (serum is squeezed from the coagulum). Clot retraction permits greater blood flow in the damaged area while the tissue is being repaired. Failures of clot retraction can be associated with reduced platelet numbers.
Clot Growth
Once blood coagulation has been initiated, the process extends into the surrounding blood; this is known as clot growth. Clot growth stops when blood flows fast enough to remove the thrombin that has been generated; this thrombin has not been otherwise absorbed by the fibrin that is formed and by the other activated factors. The thrombin and activated factors washed away by the blood are not effective because they have been diluted and because natural anticoagulant substances in plasma (e.g., antithrombin ill) are present. These substances can prevent unwanted coagulation when procoagulants (substances favoring coagulation) are present in small quantities.
Fibrin Degradation
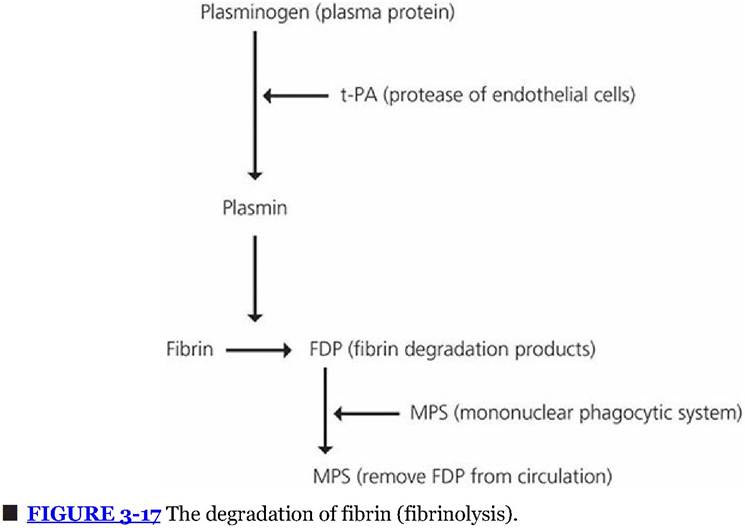
After hemostasis has been established, the damaged vascular area is repaired by new tissue growth assisted by growth factors released from activated platelets. The fibrin that was formed to assist in the hemostatic process undergoes degradation (fibrinolysis) by a proteolytic enzyme called plasmin (Figure 3-17). Plasminogen, a protein present in plasma, becomes entrapped within the clot when it is formed. Plasminogen is activated to become plasmin by substances in blood and tissues known as plasminogen activators. The principal endogenous plasminogen activator is tissue-type plasminogen activator (t-PA), which is released from endothelial cells when they are stimulated by the presence of thrombin or by stasis of blood. Plasmin degrades the fibrin molecule into protein fragments known as fibrin degradation products (FDPs). When the outer surface of the fibrin clot is removed, fresh surfaces are exposed and degraded until clot removal is complete. The FDPs, platelets, and other cell debris are removed from the circulation by the MPS. Tissue-type plasminogen activator is produced commercially for human medical use to dissolve clots that are lodged in vessels and that block blood flow (e.g., coronary arteries).
■