Microbiome and carcinogenesis
Cancer is a complex disease, in which cumulative genetic, epigenetic physiological, immunological and biochemical changes occur incessantly in the tumor tissue, contributing to the complexity of the understanding, treatment and management of the disease.
It is estimated that microorganisms could be associated with 15-20% of cancers [29].As mentioned, the microbiota has an essential role in host health, in which a beneficial relationship is established, however, dysbiotic states can trigger several diseases, including cancer. Scott and colleagues proposed that in the etiopathogen- esis of cancer, dysbiosis should be considered a persistent exit of the host microbiome from the health-associated homeostatic state (consisting of mutualists and commensals), towards a cancer promoting and/or sustaining phenotype (parasitism or amensalism) [25]. Currently, metataxonomic and metagenomics studies have documented and compared the diversity and abundance of microbes in different parts of the body between healthy and diseased patients. In veterinary medicine, it has been demonstrated a significant difference in the microbial communities in dogs with intestinal and multicentric lymphoma and with colorectal tumors comparing to healthy dogs [12, 13, 30]. However, these studies cannot distinguish whether some alterations in microbiota are causes or effects of cancer, describing only the different microbial communities found among the study groups.
The microbiome causative role has been demonstrated by controlled pre-clinical studies utilizing germfree (i.e., devoid of any microbiota) mouse models colonized with selected bacteria. For example, several family members of Enterobacteriaceae, including Escherichia coli, harbor an island of polyketide synthase (pks) pathogenicity that synthesizes a genotoxin called colibactin [31]. In an experimental study, knockout mice for IL-10 were mono-associated with two strains of E.
coli that were pks + or Δpks (with and without pks, respectively) and treated with pro- carcinogenic azoxymethane to induce colorectal tumors to demonstrate that pks play a causal role in tumorigenesis [31]. All mono-associated pks + mice developed invasive carcinoma, in contrast, none of the Δpks mono-associated mice exhibited full invasion [31]. This result suggests that the presence of E. coli pks accelerates the progression from dysplasia to invasive carcinoma through the genotoxicity of colibactin, an example of pathway of the microbiota-associated carcinogenesis process.In a recent consensus on the human microbiome role in carcinogenesis, expert opinion was that the microbiome is one apex of a tripartite, multidirectional interactome alongside environmental factors (such as diet, obesity) and an epigenetically/genetically vulnerable host that combine to cause cancer [25]. Gastrointestinal microbiome, which comprises 99% of the microbial mass, not only has the greatest both local and long-distance effects on overall health and metabolic status, but it is also the best investigated microbiome and serves as a model for understanding host-microbiota interactions and disease [32]. Due to its location, gut microbiome has been well studied as a contributor to colorectal carcinogenesis [33]. Other organs with a well-characterized microbiome include the skin and the vagina [34, 35]. The microbiome of each organ is distinct suggesting that effects on inflammation and carcinogenesis are likely to be organ specific. Although many organs (e.g. liver and brain), does not have a known microbiome, they may be exposed to pathogen-associated molecular patterns (PAMPs) and bacterial metabolites through anatomical links with the gut [32, 36].
For a better understanding of the microbiome role in carcinogenesis it is important to recognize that bacteria can be found in the tumor tissue itself, in normal adjacent tissue and in tumor sites, such as intestine and genitourinary tract, with overlap between these sites (Figure 1).
According to Picardo, the microorganisms inside, adjacent and distant from the tumor can play a role in cancer development and progression and interactions between these microbial populations together with the indirect gut microbiome effects have the potential to influence the disease development [37].At the molecular level, the mechanisms by which microorganisms can contribute to carcinogenesis are multiple and varied, which may broadly be categorized into genomic integration and genotoxicity (by a direct oncogenic effect of microorganisms and their products); promotion of immunological modifications (which disrupts host cancer immunosurveillance through the induction of pro-inflammatory and immunosuppressive pathways); and metabolic reprogramming (by altering circulating metabolites which become pro-carcinogenic and by stimulating the synthesis of trophic factors for cancer cells by the host). Many of these actions can harm the host indirectly, as microbes optimizes conditions for their survival may result in a final common pathway of prolonged host cell survival, enhanced replicative capacity and dedifferentiation [25, 33]. These mechanisms converge to hallmarks of cancer [38] and will be described in more detail below.
3.1 Genomic integration
Although the microbiome viral communities have not been studied as much as the bacterial community, the virus’s ability to integrate into the host genome is a causal mechanism of cancer both in dogs and humans.
A remarkable example is the human papilloma virus (high-risk HPV 16 and 18) and its association with human cervix cancer. The key event of HPV-induced carcinogenesis is the integration of two HPV genes (E6 e E7) into the host genomic DNA [39]. In proliferating cells of the basal layer of the uterine cervix, the viral
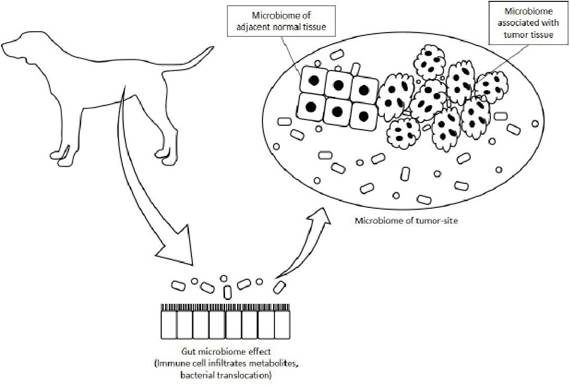
Figure 1.
The relationship between tumor and microbiomes (adapted from Picardo et al., 2019) [37].
genome persists as episomes, replicates in the suprabasal cells and can infiltrate deeper layers [40]. The HPV E6 and E7 genes are regularly present and expressed in the tumor tissue [40]. Their expression and the loss of expression of the E2 region (which negatively regulates E6 and E7) in the integrated HPV genomes cause the disruption of tumor suppressor genes that result in dysregulation of cell growth and inhibition of apoptosis [41]. Therefore, the overexpression of these viral genes synergistically acts to immortalize host cells, a cancer hallmark.
In dogs, investigations with canine papillomavirus (CPVs) have been limited to the association of different CPV genotypes with neoplastic lesions. Up to now, 20 CPVs types have been reported [42]. In skin, most genotypes of CPVs cause benign lesions, such as warts and pigmented/viral plaques or papillomas, which are selflimiting lesions such as those of oral papillomatosis [43].
Dogs that develop extensive papillomatosis may also be predisposed to oral squamous cell carcinoma (SCC) [44]. The detection of CPVs in malignant epithelial lesions is increasing in recent years [42, 45, 46]. CPV 1, 2, 3, 7, 12, 16, and 17 have been reported to cause epithelium neoplastic transformation. In a retrospective study, Thaiwong and colleagues (2018) described 7 dogs bearing benign papillomas associated with CPV1 and also the histological evidence of CPV1 causing malignant transformation of carcinoma in situ (ISC) and SCC. Later, the same group showed the expression of p53 and p16 proteins in cells infected with CPV1 in benign papillomas and lesions that progressed to SCC [42].
In a recent retrospective study, CPVs were successfully detected in 11 skin tissue samples and 4 oral tissues obtained from a cohort of canine papillomas and SCCs by PCR and through the detection of intralesional viral antigens using immunohistochemistry [46]. After sequencing, CPV 1, 2 and 6 were detected in the benign lesions, while CPV 9, 15 and 16 were detected in the SCCs, highlighting the risk of these genotypes in the induction of epithelial carcinogenesis [46].
The first report of chromosomal integration of CPV 16 into the host genome was detected in a sample of squamous cell carcinoma, raising the possibility that CPV 16 may be a potential type of high-risk canine papillomavirus [47]. However, the CPVs oncopathogenesis should be further investigated.
3.2 Genotoxicity
The gut microbiota is mainly composed of bacteria, many of which contain toxin-producing strains that can have carcinogenic effects through interfering with the cell cycle regulation, cell growth or directly damaging the host’s DNA [48]. Pathogenic bacteria strains produce protein toxins to meet their survival needs, but these bacterial defense factors perturb the host equilibrium and affect tumor suppressor genes or oncogenes and promote host genome instability [49].
Among the large number of bacterial protein toxins, two genotoxins are well known for directly affecting the host’s DNA integrity in the host organism target cells: cytolethal distending toxin (CDT), which is produced by several gramnegative pathogenic bacteria (e.g. E. coli, Shigella dysenteriae, Campylobacter jejuni, Helicobacter sp.) [50], and colibactin toxin (produced by E. coli strains); both trigger double-strand DNA breaks in host cells contributing to carcinogenesis [50, 51]. CDT exerts a pro-carcinogenic effect mainly because it presents a DNase activity. After binding to the host cell membrane, CDT suffers receptor-mediated endo- cytosis, proceeds to the endoplasmic reticulum and is translocated to the nucleus, where promotes cytotoxicity [50]. Cell CDT intoxication induces DNA damage, which results in the stopping of target cells in the G1 and/or G2 phases of the cycle and activation of DNA repair mechanisms [50]. Subsequently, normal cells that fail to repair the damage and survive the acute phase of CDT intoxication acquire the cancer hallmark of cellular senescence or undergo apoptosis via the DNA host damage checkpoint pathways [52]. This chromosomal instability supports the notion that CDT might promote tumor initiation and progression [53].
Colibactin-producing E. coli colonize frequently the colon mucosa of patients with human colorectal cancer (CRC) being implicated in carcinogenesis and tumor progression [54, 55]. This genotoxin is found in 55-67% of human colorectal cancer compared to less than 20% of controls [31, 54]. Understanding of colibactins chemical structure and biological activity is limited, but recent studies have shown that these toxins are powerful DNA-damaging agents acting via alkylation and DNA crosslinking, whose lesions activate the DNA damage checkpoint pathway and cells present signs of incomplete DNA repair, G2/M cell cycle arrest and chromosomal instability [51, 56]. In addition, colibactin also supports tumor growth by inducing a secretory phenotype associated with senescence through growth factors secretion [57].
In veterinary medicine, Feng and colleagues identified E. coli strains encoding colibactin cytotoxic necrotizing factor (CNF) in the rectal swabs and extra- intestinal samples of macaques, whose can cause clinical and subclinical diseases [58]. Genotoxins in companion animals have not been identified so far, but the fecal microbiota composition in dogs with colorectal epithelial tumors was different from that of control dogs, where Enterobacteriaceae, Bacteroides, Helicobacter, Porphyromonas, Streptococcus and Fusobacteriaceae were overrepresented in those with tumors [13]. Thus, studies are still needed to identify genotoxins produced by bacteria to help understand the carcinogenesis of canine colorectal epithelial tumors.
3.3 Immunological modifications
There is a well-defined bidirectional interaction between the immune system and gut microbiome, playing a role in the entire organism physiology [59]. The gut microbiota is essential for normal development of innate and adaptive immunity at several levels (demonstrated by studies using germ-free mice) and the immune system regulates colonization and abundance of microbiome species, as well as the response to commensal bacteria [60-63].
The host microbiota and the immune system must communicate to maintain a balance between the inflammatory response activation and the immune tolerance preservation [64]. For this, the gut bacterial population presents both a protective and harmful interface. Unlike opportunistic bacteria, other commensals, such as Bifidobacterium infantiles and Faecalibacterium prausnitzii, induce the development of regulatory T cells that prevent an inadequate immune response and protect the host against intestinal pathogens [65]. A lack of control in pro-inflammatory and anti-inflammatory bacteria (causing an imbalance between Th17 and T-regulatory cells) establishes a dysbiotic state [65, 66]. Immunological intolerance results in a loss of homeostasis that can promote a pro-neoplastic inflammatory environment through chronic inflammation, immune evasion and immune suppression [32, 67].
The gut mucosa consists of a single epithelial cell layer with intraepithelial lymphocytes that facilitates the interaction of bacterial with immune system. The epithelial line contains Paneth cells that secrete anti-microbial molecules and goblet cells that secrete mucus to lubricate the intestinal contents and protect the epithelium, while on the skin, keratinocytes regulate the microbes by secreting antibacterial peptides [68]. The lamina propria is below the mucous layer, which contains a series of other immune cells (including antigen presenting cells and CD4 + and CD8 + T and B cells). This lymphoid tissue is the most important component of body’s immune system, capable of influencing immune responses both locally and systematically [1]. Microbe is detected using pattern recognition receptors (PRRs) represented by Toll-like receptors (TLRs) and NOD-like receptors (NLR) [69].
These are widely expressed in intestinal epithelial cells, as well as in intestinal macrophages and dendritic cells. PRRs can either control the microbiota through antibacterial mediators and thus suppress cancer, or they can promote resistance to cell death - a hallmark of cancer [38].
The systemic immune system is prepared (at the epigenetic or transcriptional level) to enact a robust response in the presence of pathogenic bacteria leading to proinflammatory immune responses or to maintain a non-inflammatory state in the absence of threat [70]. A state of disruption of the delicate balance of commensal bacteria (dysbiosis), which is characterized by a less stable microbiota, increases the potential of opportunistic pathogenic bacteria growth [71]. As seen, dysbiosis can promote impaired local, loco-regional and systemic immune responses, being able to generate a profound inflammatory state, both locally and systemically. This process is outlined in Figure 2.
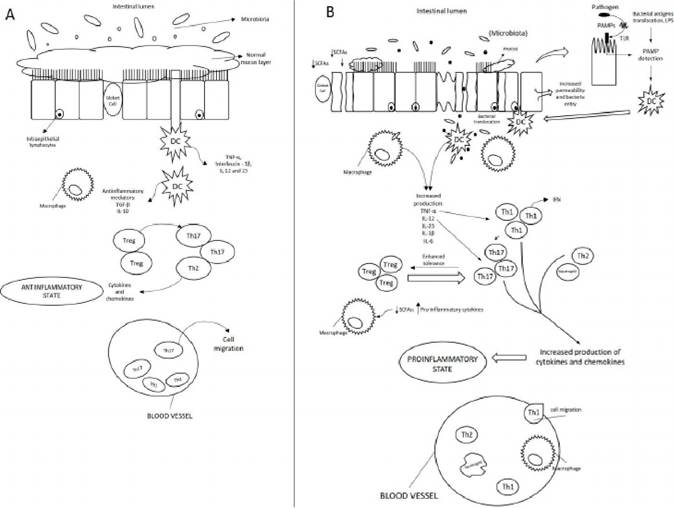
Figure 2.
The gut immune system in healthy and dysbiotic microbiome. (A): In healthy dogs, the lamina propria normally contains immune cells and secreted cytokines. These include anti-inflammatory mediators (transforminggrowth factor β [TGF-β] and interleukin (IL) -10) that down-regulate immune responses, limit excessive entry of intestinal microbiota and defend against pathogens; and noninflammatory defenses such as phagocytosis by macrophages, that assist in defending against bacteria entering the lamina propria. A homeostatic balance is maintained between regulatory T cells (Treg) and effector T helper cells (Thι, Th2, and Th17). (B): In dogs with gut dysbiosis and secondary gut inflammation, several events contribute to increased bacterial exposure, including mucus layer disruption, dysregulation of epithelial tight junctions, increased intestinal permeability, and increased bacterial adherence to epithelial cells. TLRs initiate the pro-inflammatory stimuli promoting innate local immunity through the recognition of pathogen-associated molecular patterns (PAMPs), present in bacterial antigens, such as lipopolysaccharides (LPS), peptidoglycans, flagella or unmethylated bacterial DNA CpG motifs [72]. The contact of TLRs with PAMPs initiate the innate immune response leading to secretion of cytokines and chemokines and increased expression of adhesion molecules that stimulate and facilitate specialized cells migration responsible for triggering the innate and, subsequently, the adaptive immune response (tumor necrosis factor α (TNF-α), IL-ιβ, IL-6, IL-12, IL-23, and chemokines) [73f PAMPs also induce dendritic cells (DCs) maturation that travel to mesenteric lymph nodes and present antigen to naive T cells, which differentiate into Treg and Th17 cells [68]. Tregs also contribute to intestinal homeostasis through the production of immunosuppressive cytokines, such as IL-10. Th17 cells are critical in protecting against bacterial infections because stimulates epithelial cells to secrete anti-microbial proteins and recruit neutrophils from the circulation to the gut microenvironment, resulting in a cycle of inflammation (adapted from Abraham & Cho, 2009 [68]).
In many cases, cancer development is correlated with an inflammatory host response directly to the pathogen (e.g., Helicobacter pylori and gastric adenocarcinoma) [74]. But in some cases, cancer progression may be linked to ‘sterile’ inflammatory causes that are not directly associated with infectious agents, but arising from a response to chronic uncontrolled inflammatory irritation and tissue damage, which adds to the malignant transformation [75]. In these cases, the modulatory roles in cancer development and progression are attributed to commensal or pathogenic agents. It has recently become apparent that commensal community members of microorganisms are crucially involved in tumor-promoting inflammation, which a dysbiotic state stimulates pro-inflammatory properties in the intestinal mucosa [75]. One example is intestinal lymphomagenesis associated to gut microorganism changes in host immune and inflammatory responses affecting lymphocytes [62, 66, 76-78].
The first study for microbiota-induced inflammatory tumorigenesis demonstrated that MyD88-dependent signaling controls the expression of several key modifier genes of intestinal tumorigenesis and has a critical role in cancer progression in mouse model of spontaneous intestinal tumorigenesis and in mice treated with multiple injections of azoxymethane [79]. This revealed that innate immune signaling pathway to intestinal microorganisms is an important factor in intestinal tumorigenesis [79].
It was revealed that mucosal associated invariant T (MAIT) cells from human breast ducts mediate a selective T-helper 17 cell response to human breast carcinoma cells exposed to microbial compounds [80]. This result shows that the presence of bacteria in neoplastic epithelial cells can shape the MAIT cells responses by inflammatory mediators during breast carcinogenesis [80]. Using a mouse model of cutaneous T cell lymphoma (CTCL), it was demonstrated that T cell receptor engagement is critical for the T lymphocytes malignant transformation and that disease progression is also dependent on microbiota [81].
Studies emphasize that inflammatory response to microbial commensal does not occur only in sites of direct contact between the tumor and the microbiota. It was demonstrated an increase of intestinal bacteria translocation associated to inflammation and fibrosis in human chronic liver diseases and also TLR4 activation in non hematopoietic cells in liver carcinogenesis [82]. Thus, there is evidence that intestinal microbiota can affect not only local immunity, but also systemic immune responses.
In veterinary medicine, fecal microbial communities analysis revealed significant lower bacterial diversity and distinct microbial communities in dogs with idiopathic inflammatory bowel disease (IBD) compared to healthy control dogs [30, 83]. This intestinal dysbiosis was correlated with an increase in E. coli, a group of particular interest due to its ability to stimulate inflammatory cytokines in human and canine patients with IBD [84-86]. In dogs, the fecal microbiota of patients with intestinal lymphoma, multicentric lymphoma and colorectal tumors showed a significant difference in its composition when compared to clinically healthy dogs microbiota [12, 13, 30]. However, whether these described dysbiotic states play a role in carcinogenesis remains to be determined in dogs.
3.4 Metabolic reprogramming
The metabolome is considered the link between genotypes and phenotypes [87]. It constitutes a set of metabolites synthesized by a biological system, which can be identified by recent “omics” technology called metabolomics, that allows the detection, identification and quantification of intermediate metabolism and, therefore, it can better reflect biological changes in tumorigenesis [88].
Oscillations in microbiota composition induce metabolic changes that can result in host phenotype modifications [89]. Bacterial metabolites production is one of the main signaling pathways between host and its microbiome, and metabolic reprogramming is a central feature of cancer, enabling cells to generate more energy and macromolecules for cancer cell growth, proliferation and division [90].
Microbiome-to-host crosstalk occurs by secreting bacterial metabolites and, after absorption, they enter the circulation and reach the target cells, where they exert their biological effects [91]. Microbial metabolites are detected in peripheral blood (blood metabolome) and feces (fecal metabolome) and have been identified as biomarkers of several diseases, including cancer [92, 93]. The interaction between gut microbiome and fecal and blood metabolome include several mechanisms: a) microbiome can affect gut barrier integrity and alter metabolites absorption (in this case, the same metabolite is associated with a species/pathway in the blood and feces, but the effects directions are opposite); b) direct microbiome-host cell interaction results in host systemic modulation (in this case, the species are associated with blood metabolites, but not fecal metabolites) [94]. In a metage- nomic and metabolomic study of 1,004 twins, metabolic pathways were associated with 34% of blood and 95% of fecal metabolites and it was estimated that microbiome was involved in a dialog between 71% of feces and 15% of blood metabolites, highlighting the interaction importance between microbiome and systemic and fecal metabolic environments to identify therapeutic and diagnostic targets [94].
Microbiomes of healthy subjects may share similarities in their metabolic pathways and the fecal metabolome provides a functional readout of microbial activity and can be used as an intermediate phenotype mediating host-microbiome interaction [29, 30, 95]. Zierer and associates (2018) showed that fecal metabolome largely reflects gut microbial composition and fecal metabolic profiling thus is a novel tool to explore links among microbiome composition, host phenotypes, and heritable complex traits [96].
To facilitate understanding, the most investigated bacterial metabolites and enzyme activities can be divided according to the expected effects into more protective or harmful to gut health and carcinogenesis [97].
3.4.1 Protective metabolites (tumor-suppressive metabolites)
3.4.1.1 Short chain fatty acids (SCFAs)
Fermentation of non-digestible carbohydrates from dietary fiber generates SCFAs, such as acetate, butyrate, formate, lactate and propionate [98]. The SCFAs have a key role in gut homeostasis maintenance and epithelial integrity including anti-inflammatory and antiproliferative tumor suppressive effects [98]. Butyrate has been correlated with defense against colon and liver cancer, through its well- known role in regulating inflammation and autophagy [99]. Butyrate production is associated to some Firmicutes, Eubacterium rectale, Roseburia spp., Eubacterium hal- lii, Coprococcus catus, Faecalibacterium prausnitzii [100]. It is rapidly adsorbed from gut lumen and is preferentially used as an energy source by gut epithelial cells, then its concentration in the systemic circulation is low. Butyrate is fundamental in epigenetic control; once located inside the cell, inhibits activity of histone deacetylases (HDACs) in colonocytes and immune cells, which promotes the hyperacetylation of histones, allowing transcription factors to bind to DNA and genes to be expressed [99]. This has multiple consequences for gene expression and cellular differentiation including: downregulation of pro-inflammatory cytokines (IL-6 and IL-12) in colonic macrophages; induction of differentiation of Treg cells that express transcription factor FOXP3 (crucial role in controlling intestinal inflammation); and increased acetylation results in higher expression of FOXP3 [99, 101]. As a consequence of HDAC inhibition, butyrate triggers the factor activator protein 1 (AP-1) signaling pathway in the epithelial cell lines that controls cell proliferation and apoptosis [102] (Figure 3).
SCFAs modulate several cancer hallmarks, such as cell proliferation, apoptosis and level of expression of certain genes (via inhibition of HDACs), mechanisms that lead to high anticancer activity (Figure 3). This protection can affect both stroma and cancer cells, since they have free fatty acid receptors. It was demonstrated that microbial fermentation of high-fiber diet increased concentrations of butyrate in blood and tumor and significantly decreased tumor growth in mouse with lymphoma, suggesting that dietary fiber protects against human lymphoma cancer [104]. A metabolomics-proteomics approach in colorectal cancer provided a mechanistic link between the M2 isoform of a pyruvate kinase (a direct binding target of butyrate) and metabolic remodeling and the antitumorigenic function of butyrate, highlighting an applicable approach to uncovering protein targets for small molecules with biological functions [105].
Studies in veterinary medicine are very scarce. There is one comparative study reporting higher concentrations of β-hydroxybutyrate in blood from dogs with lymphoma than in healthy dogs, but further investigations are essential to understand the significance of this increase [106]. Another research demonstrated that fecal dysbiosis in dogs with acute diarrhea was associated with altered systemic metabolic states, in which concentrations of fecal propionic acid were significantly decreased compared to healthy dogs [107]. In addition, dogs with inflammatory colorectal polyps (ICRP) showed lower amounts of propionic acid and lower proportions of Bifidobacterium compared to feces of control dogs suggesting that the association
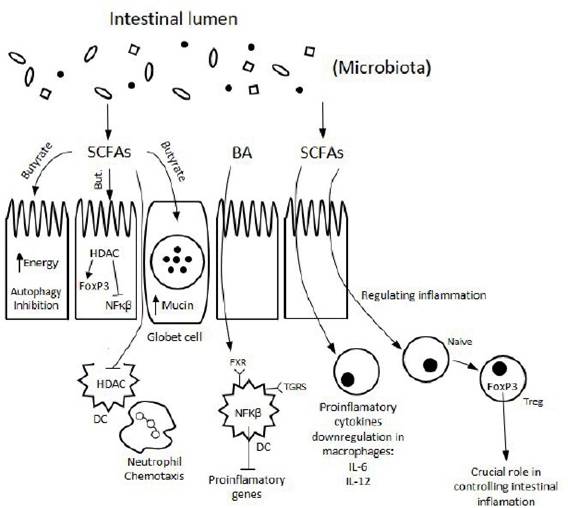
Figure 3.
Modulation of immune signaling through microbial metabolites SCFAs and BA. The metabolic effects directly stimulate the cells of the immune system or are relayed by the intestinal epithelium (adapted from Levy et al., 2019 [103].
between fecal dysbiosis and fecal SCFA concentrations may contribute to ICRP pathogenesis and therapy [108].
3.4.1.2 Phytochemicals
Phytochemicals are bioactive non-nutrient chemical compounds found in fruits, vegetables, grains, and other plant foods, which have biological effects associated with reduced risk of diseases, including cancer [109]. They can be categorized into polyphenols, organosulfur compounds, carotenoids, alkaloids, and nitrogen compounds, but the polyphenols are the most studied ones [109].
Their anti-cancer role includes antioxidant effects, modulation of xenobiotic detoxification pathways and cell proliferation, apoptosis and inflammation [110]. They neutralize reactive oxygen species (ROS) that can damage DNA and predispose to carcinogenesis [97]. A study in human breast cancer cell lines, showed that aqueous extract of the Pouteria sapota leaf is rich in phytochemicals with antioxidant properties and significant anti-cancer effects [111]. There is still need for more research and clinical trials in humans and dogs that identify and illustrate the action of phytochemicals.
3.4.2 Harmful metabolite (oncometabolite)
3.4.2.1 Bile acids (BAs)
“Primary” bile acids are synthesized from cholesterol in the liver as cholic acid (CA) and chenodeoxycholic acid (CDCA). When the gallbladder is stimulated after a meal, BA flows into the duodenum and proceeds to the ileum to be actively reabsorbed, returning back to the liver through the portal bloodstream [112]. About 15% of BAs will escape ileum absorption and enter the colon, where the resident microbiota will transform them into secondary BAs (deoxycholic acid, DCA and lithocholic acid, LCA) that have pro and anticancer activity [112]. The enzyme responsible for this conversion is 7α∕β hydroxysteroid dehydrogenase (HSDH), and it is produced specially by gram-positive Clostridium species such as Clostridium scindens [113].
Quantitative or qualitative BA pool perturbations may greatly affect several BA physiological body functions [113]. The consumption of a high-fat diet changes the gut microbiome and increases the level of DCA, that can promote carcinogenesis in colorectal and liver cancer [114, 115]. Pathways linking BAs to carcinogenesis involve the generation of ROS and reactive nitrogen species (RNS), which cause DNA damage, apoptose and epigenetic changes [112]. Moreover, BAs also exert strong antimicrobial activities, as they damage bacterial cell membranes, contributing to changes in gut microbiota (Figure 3). These mechanisms can also be secondary to environmental stimuli (particularly in the context of obesity) and their relationships with human cancer have been recognized as critical in gastrointestinal tract, prostate and breast tissues [116-118].
There are some publications covering changes in the fecal BA profile in canine chronic inflammatory enteropathy and extrahepatic congenital portosystemic shunts, but not in carcinogenesis [119-121].
4.