Diseases of the Nervous System
CNS disorders that damage areas of the hypothalamus associated with temperature regulation may lead to either decreases or increases in body temperature, although hypothermia is most common.
Hemorrhage, space-occupying masses (abscesses, tumors), infectious or inflammatory diseases, and degenerative disorders have all been implicated in hyperthermia. Hyperthermia is a common occurrence in humans with brain or spinal cord injury.40 Central hyperthermia is usually characterized by lack of diurnal variation, absence of sweating, resistance to antipyretic 3 28drugs, and excessive response to external cooling.3,
Fever
True fever differs from other hyperthermic states in that the desired core body temperature or set point is elevated.1 This
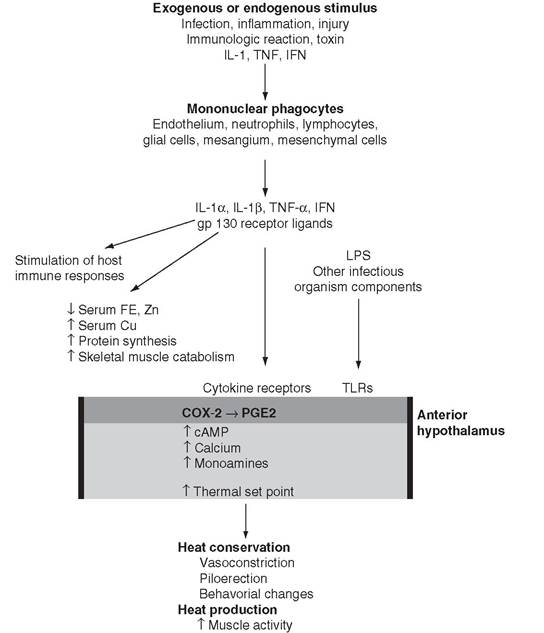
FIG. 4.2 Pathogenesis of fever. OVLT, Organum vasculosum laminae terminalis endothelium; TLRs, Toll-like receptors.
new, higher set point is vigorously defended by the same mechanisms that maintain body temperature in health. Initiation of the febrile state can occur by a variety of infectious, inflammatory, immunologic, neoplastic, or injurious conditions. In the classic model of fever, these stimuli cause the production of multifunctional pyrogenic cytokines by a wide variety of cells, but primarily by fixed or circulating monocytes and macrophages37 (Fig. 4.2). Currently at least 11 cytokines have been shown to induce the febrile response in humans and animals. Of these cytokines, interleukin 1 (IL-1α, IL-1β) and tumor necrosis factor alpha (TNF-α) are the most potent. Pyrogenic cytokines reach the POAH via the circulation and induce the production of arachidonic acid and its metabolism to prostaglandin E2 (PGE2), by the cyclooxygenase (COX)-2 pathway.
This process will eventually result in a higher “set point” within the hypothalamic thermoregulatory center41-42 (see Fig. 4.2). COX inhibitors, specifically COX-2 inhibitors, effectively reduce the febrile temperature to normal but have no effect on normal body temperature.3,42 In addition, pyrogenic cytokines stimulate the activation and proliferation of T lymphocytes and of antibody-producing B lymphocytes, which, in turn, produce additional cytokines that both enhance and inhibit further production of pyrogenic cytokines.43-44 Finally, pyrogenic cytokines, particularly IL-1 and TNF-α, cause membrane perturbation in a variety of body tissues, with a resultant increase in phospholipases and the production of arachidonic acid. Subsequent production of mediators is dependent on the metabolic pathways for arachidonic acid in the target tissue. Prostaglandins induced by pyrogenic cytokines have been shown to stimulate the muscle catabolism associated with fever and to induce collagenase synthesis from synovial cells.45 These processes contribute to the muscle and joint pain associated with fevers that are relieved by COX inhibitors.Local tissue responses to IL-1β and TNF-α may stimulate afferent neural impulses that are responsible for many of the behavioral changes (increased sleep, decreased appetite, and loss of social behavior) associated with fever.
This model, however, does not fully explain the presence of fever, because specific blockade of IL-1 or TNF-α activity does not diminish the febrile response to lipopolysaccharide (LPS) or other microbial products in experimental studies or in patients with natural infections. Microbial products from a variety of agents bind to Toll-like receptors (TLRs) on the surface of cells. TLRs and IL-1 receptors share the same signaling areas and, along with other pyrogenic cytokines, a common pathway to activate nuclear factor (NF)-κB. Activated NF-κB in turn results in expression of COX-2 or COX-3 and PGE2 synthesis.
Mice deficient in COX-2 and injected with LPS, IL-1, TNF-α, or IL-6 either intravenously or within the CNS do not develop a fever.42 Thus the induction of COX-2 and the subsequent production of PGE2 provide a common pathway for divergent pyrogens to produce the febrile response (see Fig. 4.2).There is also an apparent role for the vagal nerve in the production of the febrile response. Studies in laboratory animals in which the hepatic branch of the vagus nerve is severed have shown diminution of the febrile response to a relatively low dose of intraperitoneally injected LPS but not intramuscularly administered LPS or high-dose intraperitoneally injected LPS. Local production of cytokines may stimulate primary hepatic vagal receptors that, via vagal afferent fibers and A1/A2 noradrenergic cell groups in the brainstem, release noradrenaline, which subsequently induces the production of PGE2 and fever.46 Transection of the visceral vagal afferents has been shown to attenuate febrile behavioral responses, but not the associated temperature elevation, to high- dose intraperitoneal LPS injection in rats.47
In addition to a rise in body temperature, the febrile state is accompanied by a variety of metabolic, hematologic, and immunologic changes. IL-6 and IL-11, induced by IL-1α, IL-1β, and TNF-α, induce the synthesis of fibrinogen, C-reactive protein, haptoglobin, ceruloplasmin, and certain macroglobulins, known collectively as acute phase proteins, by hepatocytes.42 In addition, these cytokines mediate the accompanying hypoferremia, hypozincemia, and hypercupremia of the acute-phase response.
Physiologic control of the febrile response is multifactorial and prevents extreme elevations in body temperature that are incompatible with life in most instances. TNF-α inhibits further production of itself. IL-1 and other inhibitory cytokines stimulate the production of IL-1 receptor agonist, which prevents further binding of IL-1.
In addition, one of the receptors on cell surfaces for IL-1 (type II receptor) does not result in cell signaling and is thought to serve as a “decoy” receptor to decrease the concentration of IL-1.48 IL-10, induced by pyrogenic cytokines, inhibits IL-1, TNF, and IL-6 production and suppresses the production of IL-2 and IFN-γ by T-helper cells.48-49 Circulating pyrogenic cytokines may be bound to carrier molecules that reduce or prevent the interaction with receptors. For example, IL-1β has been shown to bind to α2-macroglobulin, a protein increased during the acute phase response.3 Glucocorticoids inhibit the transcription of numerous genes encoding the pyrogenic cytokines IL-1β, Il-6, and TNF-α. Within the brain, both arginine vasopressin (AVP) and alpha melanocyte stimulating hormone (αMSH) act as potent antipyretic agents. Receptors for AVP, which acts as a neurotransmitter within the brain, are found lateral to the POAH and decrease fever in both natural and experimentally induced fever, whereas injection of AVP receptor antagonist elevates fever and delays defervescence.50-51 αMSH binds to local melanocortin receptors within the brain and on cell receptors of immune cells to decrease fever and inflammation. When administered systemically to humans, αMSH is 20,000 times more potent on a molar basis than acetaminophen in decreasing fever from endogenous pyrogens.51 In the brain, nitric oxide (NO) participates in regulation of body temperature by activating soluble guanylate cyclase and increasing cyclic guanosine monophosphate (cGMP) levels. Intracerebral ventricular injection of NO donors depresses fever in rats, whereas inhibition of NO synthesis within the CNS enhances fever.48In summary, the occurrence of fever during disease results from a complex interaction of multiple cytokines and microbial products that act locally (site of tissue injury), systemically (in the circulation), and in the POAH of the brain and affect the immune, endocrine, and nervous systems.