HEPATIC DISEASES
Many diseases of the liver are treatable when a definitive diagnosis is known. However, proper management requires an understanding of the etiology, proper interpretation of diagnostic tests, and an etiologic diagnosis based on hepatic biopsy specimen analysis.
This discussion will cover the most commonly encountered diseases of the liver and review specific treatment when appropriate.Inflammatory Diseases of the Liver
Inflammatory diseases represent one of the most common manifestations of hepatic disease. The liver is often affected with infectious and toxic inflammatory diseases because it has an active reticuloendothelial cell function and plays an important role in detoxifying agents absorbed from the bowel. The liver also receives a large portion of the cardiac output and therefore has potential for systemic hematogenous involvement. The liver also may be involved in noninfectious immune-mediated reactions.
Noninfectious Inflammatory Diseases
Noninfectious inflammatory diseases are among the most common hepatic diseases seen. Unfortunately, the cause is often unclear, and they remain idiopathic entities. In addition to primary inflammatory hepatic disease, the liver can be involved secondary to disease in other organs (“innocent bystander”). Because the liver receives GI products and toxins in portal blood, primary GI disease resulting in mucosal damage can lead to increased absorption of these agents in the portal circulation. These can cause direct damage to the liver (toxic hepatopa- thy) or incite immunologic reactions leading to inflammation in the liver. It is not uncommon to find elevated hepatic enzyme activities associated with idiopathic inflammatory bowel disease (lymphocytic-plasmacytic enteritis or colitis), viral enteritis (parvovirus, coronavirus), or severe pancreatitis. In this setting, absorption of endotoxins and other toxic products results in hepatic damage and inflammation.
These cases are often referred to as a “reactive hepatopathy.” Generally there are increases in serum transaminase activities (ALT, AST) but normal concentrations of serum bile acids. In addition, the hepatic abnormalities resolve when the underlying disease is treated.Drugs can also cause hepatic damage resulting in inflammation, including anticonvulsants and
314 chapter 9 I DiseasesoftheliverandhepatobiliarySystem
certain antiparasitic drugs (mebendazole, thia- cetarsamide). Finally,metabolic abnormalities such as copper-storage diseases can result in hepatic inflammation. The histologic appearance of all these entities can be very similar. Therefore the clinician must be aware of the many factors that can lead to hepatic inflammation and thoroughly evaluate the patient to manage the disorder appropriately.
Chronic Active Hepatitis, Chronic Hepatitis
CAH is defined as an idiopathic active inflammatory disease of the liver that is chronic in duration. The term has been applied to a proposed specific disease analogous to the disease in humans of the same name or to a description of the pathologic process that results in the histologic appearance characterized by chronic ongoing inflammation. Some clinicians believe that the disorder is not a specific disease but rather a general reaction by the liver to any injury, with the histologic pattern one of nonspecific inflammation as a response to any insult. Others believe that the clinical features, histologic pattern, and response to immune- modulating drugs are similar enough to the disease in humans to warrant the name as a true disease entity.
Several distinct clinical entities can result in chronic hepatitis, including copper-storage diseases (Bedlington terrier, West Highland white terrier, Skye terrier, and possibly the Doberman pinscher), infectious diseases (viral hepatitis, leptospirosis), drugs (anticonvulsants), or idiopathic causes. This discussion will focus primarily on idiopathic causes, although it must be emphasized that diseases with a known underlying cause must be looked for and that not all cases of chronic hepatitis are “steroid-responsive” or “autoimmune.
”When an underlying cause for chronic hepatitis cannot be identified, a pathogenesis similar to that described for humans (Figure 9-1) may have merit. Briefly, it is proposed that immunologic factors lead to the perpetuation of inflammation following hepatocyte damage caused by any agent. Following hepatocyte injury, there is a release of hepatic antigens previously not exposed to the systemic immune system (hidden from immune surveillance). This results in an influx of inflammatory cells (primarily lymphocytes and plasma cells), which cause antibody-mediated and complement-mediated cytotoxicity. This results in further hepatocyte injury, and this vicious cycle is perpetuated by these immunologic events and occurs long after the initial insult is gone. At first the reaction occurs near the portal triads, but eventually it extends beyond the limiting plate of hepatocytes (the single-cell—thick layer of hepatocytes surrounding the portal triad) into the hepatic lobule. This eventually results in necrosis and bridging fibrosis (inflammation and fibrosis extending between adjacent portal areas). When the normal hepatic architecture is lost and the fibrotic process becomes diffuse, it is termed cirrhosis. Whether these events occur in the dog as has been proposed in humans, and whether the syndrome seen in dogs is analogous to CAH in human beings, is unknown.
Clinical Features of Chronic Active
Hepatitis in Dogs
CAH is represented in approximately 5% to 16% of dogs undergoing hepatic biopsy in one study. The disease occurs primarily in middle-age dogs (average 6 years), with the majority (greater than
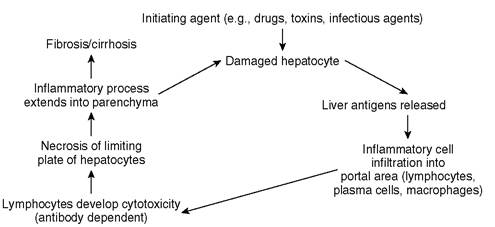
FIGURE 9-1 Pathogenesis of chronic active hepatitis.
75%) occurring in females. There is a marked predisposition in Doberman pinschers, with virtually all cases (greater than 95%) occurring in females of this breed. Because of the finding of marked copper accumulation in the livers of affected Doberman pinschers, it is unclear if the disease seen in this breed is the same as that occurring in other breeds.
However, copper is normally excreted in the bile, and it accumulates in hepatocytes with any cholestatic disorder. The finding of high hepatic copper concentrations in two Doberman pinschers with subacute hepatitis (i.e., without evidence of cholestasis) suggests the possibility of a primary copper-storage disease. For the purposes of this discussion, however, the disease seen in the Doberman pinscher breed will be considered with that seen as idiopathic CAH in other breeds because the clinical features are similar. There is also a breed predisposition in American cocker spaniels.Clinical signs of CAH include those typical of chronic hepatic disease, such as polyuria, polydipsia, weight loss, anorexia, icterus, ascites, abnormal bleeding tendency, depression, disorientation, and seizures. Some patients have a short fulminant clinical course and die within a short period of showing clinical signs. Others are presented with progressive signs of hepatic disease, although signs often wax and wane. A third group of dogs is asymptomatic when presented, with the disease identified from biochemical screening and subsequent hepatic biopsy specimen analysis.
Abnormal laboratory findings include increased serum activity of all hepatic enzymes (including ALT, AST, ALP, and GGT) in virtually all dogs. Most dogs (approximately 75%) have increased serum total bilirubin concentration (mild to marked), and many (approximately 50%) are hypoalbuminemic. Abnormalities in other chemistry values are inconsistent. Hepatic function tests (serum bile acids and plasma ammonia concentrations) are usually abnormal. Clotting times may be abnormal. These test results generally reflect the severity of the disease and stage in which it is detected. In one large series of dogs reported with chronic hepatitis from any cause, low serum glucose concentration and prolonged prothrombin time were the best predictors of early death (within 1 week of presentation). In dogs surviving more than 1 week, hypoalbuminemia was the laboratory change most predictive of shorter survival time.
The degree and severity of necrosis and fibrosis were accurate predictors of early death, and the presence of bridging fibrosis was the histologic change most predictive of shorter survival time in dogs surviving more than 1 week.Radiographic findings vary with the severity of the disease. Abnormal findings include microhe- patica (usually associated with terminal cirrhosis), ascites (associated with portal hypertension and hypoalbuminemia), and, in those cases that undergo angiographic evaluation, there are multiple acquired tortuous portosystemic shunts indicative of chronic portal hypertension.
The histologic appearance is not unique to CAH but can be seen in other inflammatory hepatic diseases. Therefore other inflammatory diseases must be eliminated to suggest the diagnosis of CAH. Histologic features include piecemeal necrosis, bridging necrosis, and active cirrhosis. Piecemeal necrosis refers to a specific pattern that is typical of CAH and characterized by periportal necrosis and inflammation occurring in an irregular fashion surrounding islands of normal hepatocytes. The majority of inflammatory cells are lymphocytes and plasma cells, although there are lesser numbers of neutrophils and macrophages. Accompanying features include bile duct hyperplasia, bile stasis, and regenerative nodules. Eventually there is deposition of fibrous tissue as a sequela to the inflammation and necrosis, eventually connecting adjacent portal triads (bridging necrosis and fibrosis). When normal hepatic architecture is lost and the fibrotic process becomes diffuse, it is termed cirrhosis. In most Doberman pinschers that have been evaluated, copper stains are strongly positive and quantitative measurements of hepatic copper concentration are high. Other breeds have not been studied as extensively for the presence of hepatic copper; however, I have seen several cases in other breeds with high hepatic copper concentrations histologically. The gross appearance of the liver can be normal in early cases.
As the disease progresses, the liver becomes shrunken, loses its normal lobular pattern, becomes discolored (brown, red, and yellow mottling), and the surface becomes irregular. Eventually the surface becomes coarsely nodular in texture, reflecting regenerative nodules occurring in a setting of terminal cirrhosis.Treatment of Chronic Active Hepatitis in Dogs
The treatment of CAH in dogs remains speculative because controlled clinical trials with large numbers of dogs have not been performed. The necroinflammatory response is usually progressive, and most dogs do not go into spontaneous remission. If cirrhosis is present, the likelihood of successfully managing the case is low. Therefore the prognosis is poor in severe cases, and most of these dogs die within several weeks to months despite appropriate treatment. This emphasizes the need for early detection.
There are three basic goals in treating dogs with CAH:
1. To arrest inflammation
2. To correct nutritional imbalances and treat hepatic encephalopathy with dietary management
3. To resolve fibrosis
Arresting Inflammation
Though there are no controlled trials using glucocorticoids to treat dogs with CAH, many clinicians have had success using them as part of the therapeutic regimen. In a large study of Doberman pinschers with CAH, however, there was little benefit seen with prednisone alone. In another study of chronic hepatitis in various breeds, dogs surviving more than 1 week that were treated with prednisone had improved survival compared with dogs not treated with prednisone. However, the varieties of hepatic diseases treated and lack of a prospective control group limit the validity of conclusions of that study. Studies in humans show that the combination of low-dose glucocorticoids and azathioprine (Imuran) is as efficacious as high- dose glucocorticoids alone and has fewer side effects. Because of these observations, the combination may be justified in dogs. I have had more success using a combination of prednisone and azathioprine than prednisone alone.
On a theoretical basis, prednisolone is preferred over prednisone in treating dogs with hepatic disease because the latter drug requires hepatic conversion to the former (active) drug. However, studies in humans with CAH have shown the two drugs to have equal efficacy and reach similar blood levels of the active form. Similar studies have not been performed in the dog, although I have found no difference between prednisolone and prednisone in treating dogs with hepatic disease.
The recommended starting dosage of prednisone is 0.5 to 1 mg/lb body weight per day. The starting dosage of azathioprine is 50 mg/m2 body surface area once daily. Once clinical remission is achieved (usually 3 to 4 weeks), the dose of prednisone is gradually tapered to a low maintenance dose (approximately 0.2 mg/lb body weight). Likewise, azathioprine is tapered to an alternate- day dosage schedule (at the original dose). I generally taper prednisone first and do not taper azathioprine until the dose of prednisone is low. This is because azathioprine is generally associated with fewer side effects and is better tolerated. A complete blood count (CBC) and platelet count should be obtained 3 and 6 weeks after starting azathioprine and every 2 months thereafter to look for potential myelosuppression. If this is detected, azathioprine should either be tapered or discontinued depending on the magnitude of the cytopenia. During the tapering process, patients are monitored with serum biochemistry profiles for relapses. Often it is difficult to differentiate the effect of prednisone on serum hepatic enzyme activities (especially ALP activity) from the effect of the disease process on serum enzyme activities. In this setting, clinical judgment is used to determine if prednisone should continue to be tapered. Serial bile acid assays and serum bilirubin and albumin concentrations also can be used to monitor the patient. If hepatic inflammation is being adequately controlled by azathioprine, the enzyme activities will decrease as prednisone is tapered. Eventually prednisone is discontinued, and later azathioprine is discontinued.
In cases with cholestasis the use of the hydrocholeretic drug ursodeoxycholic acid (ursodiol; Actigall or Urso) may be helpful. Ursodeoxycholic acid is a naturally occurring dihydroxylated hydrophilic bile acid found in small quantities in normal human bile and in larger quantities in the bile of certain species of bears. One of its uses is for dissolution of radiolucent gallstones. The proposed mechanism of action is that it alters the composition of bile, changing it from cholesterol- precipitating to cholesterol-solubilizing and dispersing cholesterol as liquid crystals in the bile, thus solubilizing the gallstones. In humans, ursodiol dissolves gallstones at the rate of approximately 1 mm/month and works best on radiolucent, noncalcified gallstones less than 20 mm in diameter. Side effects are rare (diarrhea), and there is no influence on either serum total cholesterol or triglyceride concentrations.
The exact mechanisms of its beneficial effects in inflammatory hepatic diseases remain controversial. It is believed that there is a favorable change in the bile acid pool, rendering retained endogenous bile acids less toxic by changing the bile acid pool from the more toxic hydrophobic bile acids to less toxic hydrophilic bile acids. It is also thought that ursodeoxycholic acid has antiinflammatory, immunomodulatory, and choleretic effects (promoting bile flow and decreasing viscosity of bile). The latter mechanism is believed to be mediated through the cholehepatic shunting hypothesis. This proposes that ursodeoxycholate is converted to ursodeoxycholic acid through addition of a hydrogen ion originating from carbonic acid. This leaves a bicarbonate ion to act as an osmotic draw for water to decrease viscosity and promote bile flow. Ursodeoxycholic acid has been used in the management of chronic hepatic diseases in humans, including CAH, primary biliary cirrhosis, and primary sclerosing cholangitis. Significant improvement in symptoms, laboratory parameters, and survival has been reported in many patients undergoing treatment for these diseases.
I have used ursodeoxycholic acid, either alone or in combination with other drugs, in patients with various cholestatic diseases (including dogs with CAH and cats with cholangiohepatitis). The drug is very well tolerated, and in some cases the response can be dramatic. Use of ursodeoxycholic acid should be considered as either primary or adjunctive (in combination with immunosuppressive or antiinflammatory drugs) treatment in patients with chronic liver diseases. It may be the only effective drug in patients in which glucocorticoid therapy or other immunosuppressive drug therapy is contraindicated or ineffective. Ursodeoxycholic acid is also a powerful choleretic agent that can be used to treat sludged bile and cholelithiasis. The drug is supplied in 300-mg capsules and more recently in 250-mg tablets. A safe and effective dose is 6 to 7 mg/lb/day, administered either once daily or divided twice a day. The drug should be given with food. Studies need to be performed to substantiate its efficacy and dosage in the dog and cat.
Vitamin E therapy is also recommended in many types of hepatic diseases, including CAH. Oxidative injury to the liver is now well recognized in several types of inflammatory processes. Free radicals are generated in chronic hepatitis by the injurious effects of certain drugs, toxins, or immunologic injury. These free radicals can damage cellular macromolecules via lipid peroxidation and thus participate in cellular injury and play an important role in initiating and/or perpetuating hepatic injury.Vitamin E reduces oxidant injury to hepatic tissue by providing protection from free radicals.Vitamin E is used at a dosage of 7 units/lb twice a day. Bile acids are required for fat-soluble vitamin E absorption, and, because they may be reduced with liver disease, a water-soluble formulation should be used. This type of formulation is available at most health food stores.
S-adenosylmethionine (SAMe) is a molecule synthesized by all cells and is critical to intermediary metabolism. It is especially important in the liver, where much of the body's intermediary metabolism occurs. SAMe is derived from the amino acid methionine and initiates three major biochemical pathways (transmethylation, transsulfuration, and aminopropylation). These pathways are involved with major anabolic and catabolic reactions that influence steroid hormone effects, carnitine synthesis, drug metabolism and detoxification, and hepatocyte and red blood cell membrane function. In addition, SAMe is involved in detoxification of toxins and protection against oxidative injury. The latter effect is in large part mediated through glutathione, a metabolite of SAMe through the transsulfuration pathway of metabolism. Glutathione depletion has been documented in approximately 45% of canine and feline hepatopathies.With glutathione depletion, oxidative injury is more likely to result in membrane damage and toxin accumulation, resulting in hepatocellular injury and death. It has been speculated that this could be minimized by supplementation with SAMe. Furthermore, SAMe has aminopropylation metabolites that contribute to antiinflammatory effects.
SAMe is available as a “nutraceutical” that has been studied experimentally and clinically in a variety of species. Oral use became possible when a stable oral salt in an enteric-coated tablet was developed. Nutramax Laboratories, Inc, currently markets it under the trade name Denosyl SD4. Several studies have shown the product to be safe in a variety of species, including dogs and cats.
Potential indications for usage in dogs and cats include hepatic necrosis, inflammatory disorders, cholestasis, drug-induced hepatotoxicity, copper storage hepatopathy, and metabolic disorders such as glucocorticoid-induced hepatopathy and idiopathic feline hepatic lipidosis. In these disorders administration of SAMe is meant to help minimize oxidative injury, protect against free radical damage, protect against the deleterious effect of retained bile acids, enhance bile flow, stabilize hepatocellular membranes, decrease inflammation, and aid in detoxification of endotoxins and other substances absorbed from the portal circulation. Since SAMe has such a diverse spectrum of effects,it may be helpful in a diverse group of hepatopathies. The currently recommended dose is 9 mg/lb divided twice daily for dogs and 90 mg (per cat) once daily for cats. It is rapidly absorbed and is best administered on an empty stomach to maximize absorption. The drug can be safely administered with other drugs used to treat hepatic diseases without toxicity or compromising effects of other drugs, including ursodeoxycholic acid. Controlled studies are needed to substantiate these recommendations and to determine which disorders are most likely to benefit from SAMe administration.
The optimal duration of treatment is unknown and can be quite variable. The decision to discontinue medication should ideally be based on follow-up hepatic biopsy specimen analysis. If there is evidence of ongoing inflammation, therapy should be continued. In the absence of follow-up hepatic biopsy specimen analysis, the decision to discontinue treatment may be arbitrary but should be based on clinical and biochemical information. Patients should be monitored at least monthly for relapses at first. Some patients require long-term (years), low-dose therapy (e.g., prednisone every 48 to 72 hours) to maintain remission.
Possible sources of sepsis should be examined for while these immunosuppressive drugs are being administered. This includes a culture of the original hepatic biopsy specimen, because it is not uncommon to grow bacteria as a secondary event in cases of hepatic failure due to abnormal hepatic reticuloendothelial cell function. Urinary tract infections are also common in dogs receiving prednisone and azathioprine.
It must be emphasized that there are no longterm controlled studies to support these recommendations in treating dogs with CAH. Such studies are needed and should help clarify the most appropriate treatment. Because Doberman pinschers are at increased risk of developing CAH, I recommend that a serum chemistry profile be performed every 6 to 12 months to screen for CAH in this breed so that treatment can be instituted before the development of clinical signs. Certainly owners who wish to provide the very best care should be given this option early in their pet's life.
Dietary Treatment and Correction of Nutritional Imbalances
Goals of dietary therapy are to minimize hyperammonemia, correct amino acid imbalances, and correct vitamin and mineral deficiencies. Feeding a controlled diet will also reduce the production and absorption of toxins from the small intestine, potential antigens that can worsen CAH. These therapeutic efforts are similar to those used to manage any case of hepatic disease symptomatically. These are discussed in detail in the section on management of hepatic disease.
In cases where there is excess hepatic copper documented by special copper stains or quantitative hepatic copper measurement, efforts must be made to minimize hepatic copper concentration. Drugs such as penicillamine (Cuprimine) and trientine (Syprine) are useful chelators of hepatic copper (enhancing urinary excretion). Supplementation with vitamin C (ascorbic acid) will also increase copper excretion in the urine. Supplementation with zinc acetate, gluconate, or sulfate will limit intestinal copper absorption and deposition in the liver, as well as remove copper from the liver. These drugs will be discussed in detail in the section on copper-storage diseases.
Resolving Fibrosis
Because fibrosis is a common sequela to CAH and its severity an accurate predictor of early death and shorter survival times in those surviving the initial 1 week postdiagnosis period, its management is an important part of managing cases of CAH. The drug of choice to decrease further fibrotic deposition and to dissolve existing fibrous tissue in the liver is colchicine. There are several studies in humans with alcoholic, posthepatitic, and primary biliary cirrhosis that show colchicine to have beneficial effects clinically, biochemically, histologically, and on survival. Colchicine has been used with success in cases of chronic hepatitis with fibrosis or cirrhosis in dogs. Controlled trials are needed to further substantiate these clinical observations.
The mechanism by which colchicine benefits patients with chronic hepatitis with cirrhosis is unclear, although it has antifibrotic and antiinflammatory effects. Its antifibrotic effects are primarily due to inhibition of microtubule assembly within cells by binding to the protein tubulin (thus interfering with collagen synthesis and secretion) and by stimulating collagenase activity (thus enhancing breakdown of existing collagen). Colchicine also interferes with the transcellular movement of collagen, reduces activity of hepatic collagen- processing enzymes, and inhibits proliferation of fibroblasts. Thus colchicine inhibits collagen production and increases collagenase-mediated removal of fibrous tissue. Colchicine also has many antiinflammatory properties. It has long been used as an antiinflammatory agent to treat gout in human beings. Its antiinflammatory effects include inhibition of leukocyte migration and degranulation and decreasing levels of interleukin-1. Because of these many effects, colchicine may be indicated in many diseases in which inflammation and fibrosis are prominent.
The recommended dosage of colchicine in the dog is 0.014 mg/lb body weight once daily. I generally use this drug in conjunction with prednisone, azathioprine, ursodeoxycholic acid, SAMe, and vitamin E. Side effects in the dog are minimal and are dose dependent. They include primarily diarrhea and rarely vomiting. If these side effects occur, the dose should be decreased by 25%. Ifthere is improvement, the dose can often be increased after a few weeks and is subsequently well tolerated. Side effects in humans are also uncommon and usually reversible. They include nausea, diarrhea, abdominal pain, alopecia, bone marrow depression, myopathy, neuropathy, and epistaxis. Controlled studies are needed to define the role of colchicine in managing CAH in dogs.
D-Penicillamine (Cuprimine) has had limited use as an antifibrotic agent in humans. It interferes with collagen cross-linking and maturation and has additional antiinflammatory effects. It may also be effective in lowering hepatic copper concentrations in Doberman pinschers with CAH. The dosage in the dog is 5 to 7 mg/lb three times a day. Unfortunately, the drug has a high incidence of side effects, especially vomiting and anorexia. These often preclude its use.
Feline Cholangitis/Cholangiohepatitis Complex
The term Cholangiohepatitis refers to a complex of diseases that includes inflammatory changes of the hepatobiliary system. Cholangiohepatitis is one of the most common hepatobiliary diseases of the cat. The diseases are named by the predominant inflammatory cell involved. The most common form is that of lymphocytic-plasmacytic cholan- giohepatitis. Other forms include a predominantly neutrophilic infiltration (suppurative cholangio- hepatitis), lymphocytic infiltration, and biliary cirrhosis (diffuse fibrosis of the biliary system, thought to be an end stage of a primary inflammatory disease).
Etiology
Despite this being one of the most common feline hepatobiliary diseases, the etiology remains unknown. Because the predominant inflammatory cells are usually lymphocytes and plasma cells, an immune-mediated etiology is suspected. There are also histologic similarities to primary biliary cirrhosis in humans, believed to have an immune- mediated basis. In humans there are abnormalities in cell-mediated and humoral immunity, circulating immune complexes, and antimitochondrial antibodies. These have not been documented in the cat because of the limitation of laboratory techniques for their detection. Additional evidence for an immune-mediated etiology is the marked improvement seen with glucocorticoid treatment in many cats. Although immune- mediated mechanisms may occur in many cats, there are concurrent diseases identified in some cases. These include pancreatitis, inflammatory bowel disease, bile duct obstruction, systemic infection, toxins, and cholelithiasis. In cases of suppurative cholangiohepatitis, it is suspected that bacterial infection may be an underlying cause. Escherichia coli or other bacteria are occasionally grown from cultures of hepatic biopsy samples in cases of either neutrophilic or lymphocytic- plasmacytic cholangiohepatitis, which supports this theory. It is unclear whether bacterial growth is a cause or effect of the hepatic disease. As with any cause of hepatic failure, decreased clearance of portal bacteria (such as E. coli ascending from the bowel) secondary to abnormal hepatic reticuloendothelial cell function can occur. When evaluating a suspected case of cholangiohepatitis, the clinician should make efforts to rule out other predisposing causes mentioned above. If no other causes are identified, immune-mediated mechanisms should be suspected.
Clinical Features
Cats may be affected with cholangiohepatitis at any age. The suppurative form is more commonly found in male cats and in younger cats, whereas the nonsuppurative form is more commonly found in older cats. There does not seem to be a breed predisposition, although there was a tendency seen in Persian cats in one report. Clinical signs are typical of those seen with hepatic failure in cats. These include anorexia, vomiting, depression, and weight loss. Most affected cats are icteric. Some affected cats have minimal depression and anorexia and have icterus as the main sign. Many cats are febrile. Clinical signs are often acute in onset, although some cats with the nonsuppurative form can have chronic illness. Despite these trends in presentation, none of these features reliably distinguishes the suppurative from the nonsuppurative form of cholangiohepatitis.
Laboratory findings include a marked increase in SALT activity, moderate increases in serum ALP and GGT activity, and usually hyperbilirubinemia. Concurrent bilirubinuria is also seen. Some cats have a mild nonregenerative anemia consistent with anemia of chronic disease. Ultrasound findings are nonspecific but may identify underlying diseases such as pancreatitis and extrahepatic bile duct obstruction.
Definitive diagnosis depends on histologic examination of hepatic biopsy tissue. Cytologic examination is usually not reliable for establishing the diagnosis. The disease is characterized by diffuse involvement of the liver, so random-location hepatic biopsy is adequate for obtaining representative tissue. There is usually a prominent infiltrate of lymphocytes and plasma cells, lymphocytes alone, or predominantly neutrophils in the portal triads (depending on the form of the disease). Portal triad fibrosis, bile duct proliferation, and centrilobular accumulation of bile with bile casts in canalicular areas is also frequently present. Septa of fibrous tissue with a variable lymphocytic infiltrate often link portal tracts and form circumscribed nodules of hepatocytes. It has been suggested that the disease can progress to biliary cirrhosis characterized by bridging portal fibrosis, bile duct proliferation and hyperplasia, and nodular regeneration with minimal inflammation. Aerobic and anaerobic culture of hepatic tissue with or without bile (obtained via cholecystocen- tesis) should be obtained. Culture is often positive, although it is not always clear if this is a primary or secondary event. In cats with suppurative cholan- giohepatitis, the most common organisms cultured (in descending order of prevalence) are E. coli, Staphylococcus, α-hemolytic streptococcus, Bacillus, Actinomyces, Bacteroides, Enterococcus, Enterobacter, and Clostridia. Prior treatment with antibiotics may result in false-negative cultures.
The liver in cats with cholangiohepatitis may appear large with rounded margins. The surface is often irregular and nodular, although it may also be smooth depending on the degree of nodular regeneration. The color is often a mottled red and brown with the normal lobular pattern being completely lost (reflecting distortion of the normal lobular architecture). There may be accentuated lobular markings giving a reticulated appearance (reflecting the prominent portal infiltrate).
Treatment
If an underlying associated disease is identified, this should be aggressively treated. For example, in cases of bile duct obstruction, surgical correction is usually necessary. This may involve biliary decompression if the obstruction can be relieved or rerouting of the bile duct through a cholecystoen- terostomy. Concurrent inflammatory bowel disease or pancreatitis should also be considered.
In cases of lymphocytic-plasmacytic cholangio- hepatitis, glucocorticoids are the drug of choice. Prednisolone (preferred over prednisone in cats due to the possibility of improved bioavailability of prednisolone in some cats) is instituted at a dosage of 1 to 2 mg/lb body weight twice a day for at least 1 month and subsequently tapered over 2 to 3 months when there is biochemical and clinical remission. If there is positive growth on culture of hepatic biopsy tissue, an appropriate antibiotic is given concurrently. Metronidazole (Flagyl) may also be a useful adjunct to treatment due to its immune modulatory effects. It is administered at a slightly reduced dosage of 3.5 mg/lb two to three times a day because it undergoes hepatic metabolism.
In cats that are refractory to the above immunosuppressive regimen or with the sclerosing form (biliary cirrhosis), low-dose pulse oral methotrexate in combination with prednisolone, metronidazole, SAMe, and ursodeoxycholic acid is used. A total dose of 0.13 mg methotrexate is given at 12hour intervals for a total of three doses over a 24hour period. If this dose is well tolerated at 7-day intervals (based on hematologic and clinical evaluation) and there is no biochemical improvement, the first morning dose is doubled to 0.26 mg. Some cats need the dosage interval extended to every 10 days because of GI side effects.
Treatment of the suppurative form of feline cholangiohepatitis involves use of an appropriate antibiotic based on culture results of hepatic biopsy specimens. If culture results are negative, amoxicillin-clavulanic acid (7 mg/lb body weight three times a day) or a quinolone antibiotic is usually a good choice. Metronidazole is effective against anaerobes and can be combined with the antibiotics mentioned above at a dosage of 3.5 mg/lb body weight two to three times a day. Duration of therapy often ranges from 2 to 6 months. In many cases there is only a transient response to antibiotic administration and subsequent concurrent glucocorticoid administration is often necessary. Clinical judgment is used in this situation to determine when glucocorticoids should be started. Some cases, however, are worsened by glucocorticoid administration, emphasizing the need for hepatic biopsy. Glucocorticoids therefore should only be given for the suppurative form as a last resort when antibiotics have failed.
In virtually all cases without extrahepatic bile duct obstruction, the use of the hydrocholeretic drug ursodeoxycholic acid (ursodiol; Actigall or Urso) may be helpful (see discussion of ursodeoxycholic acid in section on chronic active hepatitis). Ursodeoxycholic acid is a naturally occurring dihydroxylated hydrophilic bile acid found in small quantities in normal human bile and in larger quantities in the bile of certain species of bears. The exact mechanisms of its beneficial effects in inflammatory hepatic diseases remain controversial. It is believed that there is a favorable change in the bile acid pool, rendering retained endogenous bile acids less toxic by changing the bile acid pool from the more toxic hydrophobic bile acids to less toxic hydrophilic bile acids. It is also believed that ursodeoxycholic acid has antiinflammatory and immunomodulatory effects. Ursodeoxycholic acid has been used in the management of chronic hepatic diseases in humans, including CAH, primary biliary cirrhosis, and primary sclerosing cholangitis. Significant improvement in symptoms and laboratory parameters has been reported in many patients undergoing treatment for these diseases.
I have used ursodeoxycholic acid, either alone or in combination with other drugs, in patients with various cholestatic diseases (including dogs with CAH and cats with cholangiohepatitis). The drug is very well tolerated, and in some cases the response can be dramatic. Use of ursodeoxycholic acid in feline cholangiohepatitis complex should be considered as adjunctive (in combination with immunosuppressive or antiinflammatory drugs or antibiotics) with any of the histologic forms of the disease. Ursodeoxycholic acid is also a powerful choleretic agent that can be used to treat sludged bile and cholelithiasis. The recommended dose is 6 to 7 mg/lb/day, administered either once daily or divided twice a day. The newer 250-mg tablet form of the drug (Urso) has made dosing more convenient. The drug has minimal side effects, and in my experience it may dramatically improve therapeutic benefit. Studies need to be performed to substantiate its efficacy and dosage in the dog and cat. The use of SAMe also appears helpful for this disease. See section on CAH for detailed description of this drug.
Specific dietary management is generally not critical to a successful outcome; however, nutritional support may be necessary. It is more important that the affected cat eat any balanced diet than any specific diet. A protein-restricted diet is rarely necessary in cats. In most cases, response to therapy is rapid and tube feeding is not necessary. If there is prolonged anorexia, nutritional support should be provided by using an enteral feeding tube (percutaneous endoscopic gastrostomy [PEG] tube, esophagostomy tube, or nasoesophageal tube). Use of feeding tubes is described in Chapter 12.
Vitamin E therapy is also recommended in many types of hepatic diseases, including feline cholangiohepatitis. Oxidative injury to the liver is now well recognized in several types of inflammatory processes. Free radicals are generated in chronic hepatitis by the injurious effects of certain drugs or toxins or by immunologic injury. These free radicals can damage cellular macromolecules via lipid peroxidation and thus participate in cellular injury and play an important role in initiating and/or perpetuating hepatic injury. Vitamin E reduces oxidant injury to hepatic tissue by providing protection from free radicals.Vitamin E is used at a dosage of 100 to 200 units/day. Because bile acids are required for fat-soluble vitamin E absorption and may be reduced with liver disease, a water-soluble formulation should be used. This type of formulation is available at most health food stores.
The prognosis is fair to good unless the disease is histologically advanced. Many patients respond dramatically to glucocorticoid administration, and most can eventually be weaned off medication. However, some cats will relapse and need ongoing or intermittent treatment. Some cats require prednisolone at a dosage of 0.5 to 0.7 mg/lb/day on a long-term basis (months to years) for successful control of the disease process. Fortunately, the long-term side effects of glucocorticoid administration are minimal in the cat.
Hepatic Necrosis and Toxic Hepatopathies
Hepatic necrosis often leads to acute hepatic failure. Because the hepatocyte is exposed to an extensive portal and systemic venous circulation, it is susceptible to injury by a variety of etiologic agents. Hepatic necrosis can occur secondary to other hepatic processes such as inflammation or neoplasia, can be associated with known hepato- toxins, or can occur when no other cause is known. Causes of hepatic necrosis are listed in Box9-11.
Clinical Features
Clinical signs of hepatic necrosis depend on the degree of severity. Many patients are asymptomatic, with disease detected only by biochemical screening (such as many cases of hepatic trauma), whereas other cases have acute fulminant hepatic failure. In the latter instance, affected patients range from profoundly depressed to comatose, with the degree of hepatic encephalopathy depending on the cause and severity. Vomiting, anorexia, and fever are often seen. Icterus is often seen when there is periportal involvement. The presence of coagulopathies such as DIC reflect the degree of severity and usually manifest with GI bleeding, hema- temesis, ecchymoses, and excessive bleeding at venipuncture sites.
Laboratory findings include profound increases in SALT and SAST activity. Increased SALT activity correlates with histologic findings only in the initial period of hepatocellular injury, after which serum enzyme activity may persist or decline in the following days despite the persistence of extensive necrosis. The activities of serum ALP and GGT and serum bilirubin concentration are variable and reflect the degree of cholestasis. The degree of clinical severity is often poorly correlated with measurements. Other variable
BOX 9-11
Causes of Hepatic Necrosis
Chemicals
Drugs
Aflatoxins
Septicemia
Pancreatitis
Inflammatory bowel disease
Viral agents
Inflammatory hepatic disease (CAH) Systemic hypoxia
Anemia
Ischemic injury
Excessive copper storage
Heartworm-associated (postcaval syndrome) Trauma
CAH, Chronic active hepatitis. abnormalities include hypoglycemia and abnormal clotting parameters. Hepatic function tests such as serum bile acids and plasma ammonia concentration are often normal unless there is massive hepatic necrosis and hyperbilirubinemia. Laboratory abnormalities usually overlap those of other diseases such as CAH and primary hepatic neoplasia.
Histologic findings reflect the degree of hepatic necrosis. The pattern of necrosis is differentiated from that seen in CAH by its location within lobules, by the appearance of neutrophils that enter to phagocytize cellular debris, and by the absence of lymphocytes and plasma cells as the predominant inflammatory cells.
Treatment involves withdrawal of known hepa- totoxins or treatment of underlying conditions listed in Box 9-11 that are associated with hepatic necrosis. Efforts must be made to look for these disorders associated with hepatic necrosis. Additional treatment measures are aimed at providing optimum conditions for hepatic regeneration and preventing secondary complications of hepatic disease. These measures are discussed in detail in the section on management of hepatic disease.
Approach to Nonspecific Increases in Hepatic Enzyme Activities
Many patients will be detected as having increased serum activities of hepatic enzymes on routine biochemical screening or when presented with clinical illness. The clinician must try to determine if an underlying nonhepatic cause is present, including endocrinopathies (hyper- adrenocorticism, diabetes mellitus, and hyperthyroidism) and those disorders listed in Box 9-11. If no obvious underlying cause exists, the clinician must determine whether clinical signs can be attributed to hepatic disease. If these signs are serious enough to cause illness, further work-up with hepatic function tests (bile acids or blood ammonia concentration), ultrasound examination, and/or hepatic biopsy is warranted. If clinical signs are absent or mild, a baseline hepatic function test (generally serum bile acids assay) is run and is used as a monitoring parameter. Unless hepatic function is significantly abnormal, a repeat chemistry profile and hepatic function test are run in 4 to 8 weeks. If these tests are persistently abnormal, hepatic imaging and biopsy are warranted, even in patients that remain clinically normal.
Drug-Induced Hepatic Disease
Many drugs have been reported to cause hepatic disease in patients. The most common ones are listed in Box 9-12. Several types of hepatopathies are associated with drug administration, including hepatocellular necrosis, cholestasis, CAH, vacuolar changes (including steroid hepatopathy), and a combination of these processes. Hepatotoxic drugs can be further classified into those causing predictable hepatic damage (intrinsic toxicoses) and those that are idiosyncratic in their potential to cause hepatic damage. Those drugs causing predictable hepatic damage have a high incidence of hepatotoxicity and are usually dose dependent, and their effects can be reproduced in experimental animals. On the other hand, idiosyncratic reactions are characterized by occurring in a small percentage of patients, are are usually not dose dependent, and are to experimentally reproduce. Idiosyncratic toxicosis is the result of an unusual susceptibility of an affected patient to an adverse reaction resulting from metabolic aberration, hypersensitivity, or immune-mediated events. Specific mechanisms of injury are usually unknown when idiosyncratic toxicosis occurs. In general, treatment of drug-induced hepatic disease involves withdrawal of the drug and supportive care. This
BOX 9-12
Drugs Known to Cause
Hepatic Disease
Acetaminophen
Anabolic steroids
Anticonvulsant drugs (phenobarbital, primidone, phenytoin)
Antineoplastic drugs (methotrexate,
L-asparaginase, 6-mercaptopurine)
Arsenicals (thiacetarsamide)
Carprofen
Diazepam
Diethylcarbamazine
Furosemide
Glucocorticoids
Griseofulvin
Inhalation anesthetics (halothane,
methoxyflurane)
Itraconazole
Ketoconazole
Lomustine (CCNU)
Mebendazole
Mitotane (o,p-DDD)
Sulfonamides
Tetracycline
Trimethoprim-sulfadiazine discussion will concentrate on the most important drug-induced hepatopathies.
Anticonvulsant Drug-Induced Hepatic Injury
Hepatobiliary disease associated with the administration of many anticonvulsant drugs has been described, including phenytoin, primidone, and phenobarbital (either alone or in combination). These drugs often result in elevations in hepatic enzyme activities, but most patients are asymptomatic and tests of hepatic function, including serum bile acids and plasma ammonia concentrations, are often normal. However, significant hepatobiliary disease occasionally develops in patients given seemingly safe doses or when drug doses are increased to toxic levels to maintain seizure control. Toxic blood levels can also develop in patients receiving appropriate doses when hepatic failure from other causes occurs, because these drugs are metabolized and cleared by the liver. Primidone is more hepatotoxic than phenobarbital.
Clinical signs include those typical of hepatic disease, including depression, anorexia, and weight loss, with subsequent development of jaundice and other features typical of hepatoencephalopathy and end-stage hepatic disease. Terminal events often include excellent seizure control because the anticonvulsant drugs are poorly metabolized and thus achieve high blood levels. By the time clinical signs are noticed, hepatic disease is usually advanced (often with the presence of cirrhosis) and the prognosis is poor. However, it has been estimated that clinical signs develop in only 6% to 15% of dogs receiving anticonvulsant drugs long term.
Laboratory abnormalities include variable increases in SALT, SAST, and serum ALP and GGT activities. These changes are most marked in dogs receiving primidone and/or phenobarbital. Increase in SALT activity is usually reversible upon withdrawal of the drug and does not always correlate with morphologic evidence of hepatocellular necrosis. Increase in serum ALP activity is related to increased hepatic synthesis. Because these enzyme abnormalities occur in many asymptomatic dogs without significant hepatic injury, other tests of hepatic function should be used to determine whether there is impending hepatic failure, including serum bile acids, albumin, and plasma ammonia concentrations. Increased total bilirubin concentration and decreased albumin, BUN, glucose, and cholesterol concentrations, although not specific, are common indicators of hepatic failure. If these tests are abnormal, hepatic biopsy or decreasing the dosage of the anticonvulsant may be warranted.
Two distinct forms of hepatotoxic injury are related to anticonvulsant drug treatment. One form is characterized by the development of clinical signs after extended periods of treatment. Histologic findings include diffuse fibrosis, nodular regeneration, and various amounts of necrosis, lipidosis, and inflammation that eventually lead to macronodular cirrhosis. The second form of hepatotoxic injury is metabolic hepatic failure with intrahepatic cholestasis that is distinct in historical, clinical, and histologic features from those associated with cirrhosis. There is a conspicuous absence of a necroinflammatory response versus other forms of drug toxicity or acquired canine hepatic disease unrelated to drug administration.
The prognosis is poor when histologic lesions are severe and hepatic failure has occurred. Treatment involves withdrawal of anticonvulsant drugs if possible or use of alternative anticonvulsant drugs such as potassium bromide. There are no hepatotoxic effects associated with potassium bromide. There is no indication for the use of glucocorticoids unless there is an active inflammatory component. The use of colchicine might be indicated if fibrosis is a prominent feature (see section on chronic active hepatitis). The use of ursodeoxycholic acid may also be indicated due to the presence of a significant cholestatic component in most cases. The use of SAMe may also be helpful. It should be emphasized that caution should be used when attributing abnormal hepatic function and hepatic failure to anticonvulsant administration because the incidence of this problem is low. Other laboratory and ancillary tests, including hepatic biopsy specimen analysis, may well be justified in these patients.
Carprofen Toxicity
Carprofen (Rimadyl) is a commonly used nonsteroidal antiinflammatory drug (NSAID) to treat canine osteoarthritis (degenerative joint disease). It is estimated that the incidence of severe hepato- toxic reactions from carprofen is 1.4 dogs per 10,000. In one report 21 dogs were described to have hepatocellular toxicosis associated with administration of carprofen. At the time of the report, over 500,000 dogs had received carprofen. No dog in this report had evidence of a previous significant hepatopathy or medical problems predisposing to a hepatopathy. Various other drugs were given to some dogs, with no apparent relationship to developing hepatic toxicosis. Of the 21 dogs, 13 were Labrador retrievers. Dogs ranged from 4 to 15 years old (mean, 9.4 years). Carprofen was administered for alleviation of signs of musculoskeletal pain in all dogs. The amount of carpro- fen administered ranged from 0.71 to 1.41 mg/lb of body weight (mean, 1.06 mg/lb) orally every 12 hours. The duration of treatment ranged from 3 to 180 days (mean, 31 days). Clinical signs of toxicosis were noticed for 18 dogs between 5 and 30 days (mean and median, 19 days) after initiation of carprofen. Two dogs received carprofen for 60 and 180 days before developing clinical signs. One dog received the drug for 54 days and did not have clinical signs of toxicosis. The drug was discontinued after discovery of hepatic necrosis on a biopsy specimen obtained for evaluation of possible metastatic cancer in this dog. All Labrador retrievers developed clinical signs at an interval of
14 or more days after initiation of carprofen administration (mean and median, 20 days). One of these dogs received carprofen for only 3 days, but clinical abnormalities developed 18 days after the first dose.
Clinical signs associated with toxicosis were predominantly anorexia (17 dogs) and vomiting (16 dogs). Other signs noticed less frequently were lethargy, diarrhea, polyuria, polydipsia, and hematuria. Physical examination revealed icterus in
15 dogs and ascites in 1 dog. The most common laboratory abnormalities were increases in serum activities of ALT (21 of 21 dogs), AST (14 of 15 dogs), ALP (20 of 21 dogs), and serum bilirubin concentration (18 of 21 dogs). Hypoalbuminemia was seen in only 4 of 21 dogs. In addition, urinalyses were performed in 9 dogs. In 7 dogs evidence of renal disease was present (including isosthenuria with azotemia, glucosuria, proteinuria, and evidence of epithelial cells and granular casts). Hemogram, radiographic and ultrasonographic abnormalities were minimal. Histopathologic evaluation (performed in 18 of 21 dogs) revealed varying degrees of vacuolar change, ballooning degeneration, necrosis of hepatocytes, bridging fibrosis, mixed-cellular inflammation, and accumulation of bile pigment.
Fifteen of the dogs were treated. Of these dogs, 12 were hospitalized. These 12 dogs received intravenous fluids and antibiotics. Drugs used to manage GI signs included histamine H2-receptor antagonists, metoclopramide, sucralfate, and misoprostol. Three dogs were given ursodeoxycholic acid for 14 to 60 days.
Four dogs died or were euthanized within 3 to 5 days after initial examination. One dog with severe hepatic and renal failure also had perforation of the GI tract and diffuse intestinal ulcers documented during necropsy. It is unknown whether the GI tract ulcers and subsequent perforation were directly attributable to carprofen use or indirectly attributable to hypoperfusion, ischemia, or uremia. The other 17 dogs fully recovered from drug-induced hepatic disease. All 13 Labrador retrievers recovered from the hepatic injury. The mortality rate for the other breeds was 50%. For surviving dogs, vomiting resolved 1 to 5 days after supportive care was instituted and carprofen was discontinued. Inappetence was the primary persistent clinical sign, which resolved 6 to 20 days after carprofen was discontinued. Carprofen administration was discontinued but was repeatedly reinstituted and discontinued during a 1-month period for 1 dog. Clinical signs resolved after discontinuance and reappeared in association with drug administration. Laboratory evaluations were performed on all surviving dogs 3 to 4 weeks after onset of clinical signs. All dogs were markedly improved with regard to hepatic variables. Values determined 3 months after diagnosis of the toxic condition for 8 dogs were within reference ranges or only slightly increased. Fifteen of 17 surviving dogs were healthy 60 days after the episode of toxicosis. The other 2 dogs had unrelated problems.
The results of this study suggest that the drug reaction to carprofen is idiosyncratic and hostdependent in nature. Progression of the condition did not appear to correlate with the dose of carprofen, magnitude of hepatic enzyme activities, or histopathologic severity of hepatic lesions. It is also noteworthy that renal lesions were detected in a number of dogs, a well-documented side effect of other NSAIDs. Although prescreening hematologic and serum biochemical analyses may not yield results that can be used to predict dogs that will have adverse reactions to carprofen, evaluation of renal and hepatic function before administration of the drug is recommended. Dogs with renal and hepatobiliary abnormalities may be poor candidates to receive this drug, or extra caution should be used if carprofen is to be used in these dogs. In addition, serum biochemistry analysis should be obtained approximately 3 to 4 weeks after starting carprofen to detect patients with developing hepatic or renal disease. Owners should be informed of the clinical signs of drug intolerance and instructed to immediately discontinue the drug if these signs develop.
Mebendazole-Induced Hepatic Disease Mebendazole (Telmintic) is an anthelmintic drug that is useful for its effects against ascarids, hookworms, and whipworms. Though generally considered to be a safe drug, acute hepatic necrosis associated with mebendazole administration to dogs has been reported. In addition to this report, 45 additional cases of adverse drug reactions associated with mebendazole administration have been reported to the U.S. Food and Drug Administration. Based on these reports and extensive safety studies in normal animals and in induced hepatic disease, it is unclear whether mebendazole is an intrinsic (predictable) or idiosyncratic hepatotoxin. The fact that toxicity is of low incidence, difficult to reproduce experimentally, and apparently not dose related suggests that mebendazole is an idiosyncratic toxin, whereas the presence of toxicity in several members of one kennel suggests that mebendazole is an intrinsic hepatotoxin.
Regardless of the mechanism of toxicity, I recommend that mebendazole not be used. Safer and more effective anthelmintics with a similar antiparasitic spectrum such as fenbendazole (Panacur) or febantel (Drontol Plus) are recommended.
Copper-Storage Hepatopathy
Pathophysiology
The abnormal accumulation of copper in hepatocytes as a result of a metabolic defect in copper metabolism has been documented in the Bedlington terrier, West Highland white terrier, Skye terrier, and possibly the Doberman pinscher breed. These disorders are similar (but not identical) to Wilson's disease in humans. Once excessive copper accumulates in hepatocytes, it results in progressive hepatocyte destruction. The disease in these breeds must be distinguished from other causes of secondary hepatic copper accumulation. Copper is normally excreted through the biliary tract. Therefore copper can accumulate in the liver with any cholestatic disorder, including CAH or cirrhosis. In dogs with a primary copper- storage disease, copper accumulates in the liver before the development of hepatic damage or cholestasis.
Once copper is ingested, 40% to 60% is passively absorbed in the proximal small intestine, with the remainder lost in the feces. Some ingested copper is bound to the copper transport protein, metallothionein. This portion is lost in the feces. Unbound copper is absorbed from the intestine and enters the portal circulation, where it is bound to albumin and another copper transport protein, transcuprein. Copper is then transported to the liver. Within hepatocytes, copper is bound to cytosolic metallothionein and stored in lysosomes. Copper can then undergo two fates: it can be excreted in bile or bound to the copper transport protein ceruloplasmin for transport to peripheral tissues. Of these steps, the most important step that regulates copper homeostasis is biliary excretion. In dogs with abnormal copper storage, copper accumulates within the lysosomes of hepatocytes. While in the lysosomes, copper is innocuous. Once the lysosomal storage capacity is exhausted, copper breaks into the cytoplasm, where it is toxic to the hepatocytes. Excessive hepatic copper can alter hepatic membrane permeability and interfere with normal hepatocyte transport of proteins and triglycerides, and eventually these hepatocytes undergo cellular lysis and necrosis. This can result in massive release of copper from damaged hepatocytes, which when taken up in the circulation can lead to a hemolytic crisis.
In Bedlington terriers the disease is an autosomal recessive disorder. The specific defect in copper metabolism is thought to be excessive copper binding by an abnormal metallothionein in the liver, which sequesters copper in the liver (lysosomes) and reduces biliary excretion. The excess copper accumulation can be detected as early as 6 months of age and progresses with time. It is unclear whether a subset of Doberman pinschers with CAH have a primary copper-storage disease or whether the abnormal hepatic copper concentration is secondary to the cholestasis associated with the active hepatitis. One report, however, documented increased hepatic copper concentrations in two Doberman pinschers with subacute hepatitis in which cholestasis was not present histologically, leading to the speculation that a genetic defect in copper metabolism might be the primary cause of hepatic inflammatory disease in some Doberman pinschers. The disease in West Highland white terriers differs from the copper-storage disease in Bedlington terriers by comparatively lower concentrations of hepatic copper.West Highland white terriers can generally tolerate up to 2000 μg∕g (ppm) of copper.West Highland white terriers rarely accumulate excess copper throughout their lifetime. By 6 months of age a West Highland white terrier has reached its upper limit of excess copper, and some dogs with excess hepatic copper will return to normal by 1 year of age. In the Skye terrier excessive copper accumulation in the liver appears to be related to cholestasis, thought to result from a disorder of intracellular bile metabolism and abnormal bile secretion.
Although Wilson's disease in humans is similar to the disease in affected Bedlington terriers, there are several important differences.Wilson's disease often leads to copper accumulation and subsequent damage in other tissues, including the brain. These manifestations are not seen in the dog. In addition, the concentration of ceruloplasmin in humans with Wilson's disease is low, whereas the concentration of unbound copper is variable but may be high. In affected Bedlington terriers, serum copper and ceruloplasmin concentrations are normal.
Clinical Features
The disease is an autosomal recessive inherited defect. In studies in which large numbers of Bedlington terriers have been screened by liver biopsy for abnormal copper storage, approximately 50% to 80% of dogs have been affected. Recent genetic studies by VetGen, LLC, suggest that only 30% of Bedlington terriers tested are homozygous clear of the disease, 39% are homozygous affected, and the remainder (31%) are heterozygous carriers.
The copper-storage disease of Bedlington terriers can be categorized into three general groups. In the first group, affected dogs are usually young adults. They have a short, fulminant course of acute hepatic necrosis and failure with a high mortality rate. These dogs are usually icteric, anorectic, and vomiting, and they may undergo a hemolytic crisis because of copper toxicity from rapid lysis of hepatocytes. Most dogs die despite supportive measures. Sometimes a stressful event such as whelping or showing precipitates the onset of signs.
In the second group, affected dogs are usually middle-age or older. There is usually an insidious deterioration of their general condition, characterized by chronic weight loss, anorexia, intermittent vomiting, and a general unthriftiness. On presentation many dogs have hallmarks of chronic end-stage hepatic disease, including icterus and ascites.
In the third group, affected dogs are asymptomatic and the disease is detected by biochemical screening (usually with increased SALT activity) and documented by hepatic biopsy specimen analysis. It is thought that dogs in this group represent a prestage of the first two groups. In affected dogs of all groups, hepatic copper concentration can be elevated as early as a few months of age. Progressive increases in copper concentration usually occur until 5 to 6 years of age (if the patient survives), at which time levels slowly decline, although they never completely return to normal.
The most consistent laboratory abnormality is increased SALT activity, usually occurring once hepatic injury has taken place. The SALT activity usually correlates with the severity of the disease histologically, although enzyme depletion may occur with terminal cirrhosis. Serum ALP activity and serum bilirubin concentration are variable, reflecting the degree of cholestasis. A presumptive diagnosis should be considered in any Bedlington terrier with increased SALT activity, although the disease needs to be confirmed with hepatic biopsy specimen analysis. It must be pointed out that a normal SALT level does not rule out the disease, and Bedlington terriers get other forms of hepatic disease.
The diagnosis is confirmed with quantitative measurements of hepatic copper from hepatic biopsy specimens. Normal hepatic copper concentrations range from 91 to 377 μg∕g (ppm) of liver on a dry weight basis, although there is marked variability among dogs of various breeds. Formalin-fixed hepatic tissue is suitable for quantitative measurement. Dogs having values above this range are considered affected, and most affected dogs have hepatic concentrations from 5 to 50 times above normal. The disease can also be documented by histochemical staining for copper with rubeanic acid, Timm's, rhodanine, or orcein stains (most pathologists use rubeanic acid). In affected livers, granules of copper can be seen with these stains and are qualitatively estimated. The measurement of copper-64 excreted in stool following intravenous injection has also been advocated as a noninvasive method of detecting affected dogs. Histologic findings vary from normal (with the exception of excess copper accumulation) to varying severities of chronic hepatitis. The disease progresses from focal hepatitis and necrosis to features identical to those described for CAH. Eventually the disease progresses to micro- nodular and macronodular cirrhosis. The gross appearance of the liver reflects the histologic severity, ranging from normal to a fine or coarse nodular surface, with regenerative nodules reflecting end-stage liver disease.
Recently VetGen, LLC, began offering a genetic test for copper toxicosis of the Bedlington terrier breed. This test uses a linked marker that has two alleles, or marker types, called 1 and 2. It was found that over 90% of dogs that were 1/1 marker type were homozygous normal (clear of the disease) and over 90% of dogs that were affected with the disease were 2/2 marker type. Most 1/2 dogs are carriers with the 2 allele usually associated with the copper toxicosis disease allele. The finding of such a strong genetic disequilibrium allows this to be potentially a valuable test. In interpreting the results, if the dog is a 1/1, it is more than 90% likely that it is homozygous normal (clear of disease). If the dog is a 2/2, it is 72% likely that it is affected (over 90% of affected dogs are 2/2, but 72% of 2/2 are affected; 24% are carriers). If the dog is a 1/2,VetGen data indicate the dog has a 95% chance of being a carrier. This test may be helpful in making recommendations to breeders. If only 1/1 and 1/2 dogs are chosen for breeding, the 2 gene could be eliminated in subsequent generations. However, breeders should still allow liver biopsy specimens to be obtained in 1/1 dogs to be used for breeding for the near to intermediate future because it is currently the only way to detect the small number of affected dogs associated with the 1 allele. VetGen provides a collection kit for DNA using a soft cheek brush. This test can be completed before puppies are purchased at an early age.[*****]
Treatment
Early detection is essential. Therefore, because of the high incidence in the breed, it is strongly recommended that Bedlington terriers undergo biochemical screening two to three times per year. Ideally a liver biopsy should be performed at 1 year of age. Once a positive diagnosis is established, treatment depends on the stage of the disease. In affected dogs, specific therapy involves the administration of drugs that chelate copper and increase urinary excretion, as well as efforts to decrease copper absorption. Traditionally, the drug of choice has been D-Penicillamine (Cuprimine). The recommended dosage for dogs is 4.5 to 7 mg/lb two times a day. The drug should ideally be given before meals to maximize its effect. In this manner, penicillamine will remove approximately 1000 qg/g (ppm) of copper per year.When given with meals the efficacy decreases by approximately half. Common side effects include anorexia and vomiting. Further dividing the dosage into three to four daily doses and/or administering it with food often minimizes these signs. Unfortunately, these side effects may be intolerable in some dogs and necessitate discontinuation of the drug. It usually takes several years for hepatic copper concentrations to decrease to normal, and therapy must be continued for life. In addition to chelating copper, D-Penicillamine has antifibrotic properties, stabilizes lysosomes, and has immune-modulating effects that might also be of benefit in managing this disease.
More recently tetramine cupruretic agents (2,2,2-tetramine; 2,3,2-tetramine) have been evaluated and have been shown to be effective decoppering agents, lowering hepatic copper and increasing urinary excretion. These drugs are also better tolerated than D-Penicillamine. Trientine (Syprine; 2,2,2-tetramine) is my drug of choice. It has cupruretic effects similar to those of D-Penicil- lamine (i.e., removing approximately 1000 μg∕g [ppm] of copper per year), although it may attack a different copper pool. The recommended dose is 7 to 14 mg/lb two times a day. Side effects are minimal in dogs compared with those caused by D-Penicillamine. Trientine is often used first or in patients that experience side effects with D-Penicil- lamine. Trientine is not always readily available, but many pharmacists will order it or it can be ordered directly from the manufacturer (Merck). 2,3,2-Tetramine is an experimental drug that has been shown to be a potent copper chelator. It is not yet commercially available.
Additional measures to reduce hepatic copper concentrations include supplementing the diet with zinc (0.7 to 1.15 mg/lb zinc gluconate three times a day, 0.3 mg/lb zinc sulfate three times a day, or 100 mg elemental zinc as zinc acetate two times a day). Zinc induces increased concentration of intestinal metallothionein, which then binds ingested copper to intestinal epithelial cells, thus preventing copper absorption. As these cells are sloughed, copper is subsequently lost in the feces. In addition, zinc will enhance removal of copper from hepatocytes. Zinc lowers hepatic copper indirectly by affecting multiple areas of copper equilibria and by displacing copper in target tissues. Zinc also induces hepatic metallothionein, which will then bind to and sequester excessive copper into an innocuous form compared with free copper. The rate of removal of hepatic copper is relatively slow. For this reason dogs with severe or fulminant hepatitis secondary to copper accumulation are not candidates for zinc therapy alone. For these patients zinc is commonly combined with a chelating agent such as trientine. One study demonstrated marked improvement in hepatitis and hepatic copper concentrations in three Bedlington terriers and three West Highland white terriers treated with zinc acetate as the sole decoppering agent. The advantages of zinc for treatment include efficacy, low cost, and minimal side effects. With any of the above types of zinc supplements, it is important to measure serum zinc levels. The goal is to achieve plasma zinc concentrations of 200 to 600 μg∕dl. After a 3- to 6-month loading period, the dose is decreased to approximately half of the original dose. Serum zinc levels are then measured every 4 to 6 months. If the serum concentration drops below 150 μg∕dl, the dose is increased to the original dose. To be effective, zinc must be given separately from food by at least 1 hour because some food constituents such as phytates can bind zinc and diminish its efficacy. If zinc causes vomiting, it may be mixed in a tablespoon of tuna fish (in oil) to minimize nausea.
Vitamin C also might be useful because it decreases copper absorption and increases copper excretion in the urine. In addition, dogs with hepatic insufficiency are deficient in ascorbic acid. Ideally vitamin C should be given with meals. The recommended dosage is 12 mg/lb/day. Dogs should also be fed a diet low in copper concentration. Some commercial diets low in copper include Hill's Prescription Diet l∕d, Purina Fit & Trim, Purina HiPro,Wayne,ANF, Pedigree, Nutro Natural Choice, and Precise. Diets high in copper include Iams Eukanuba, Science Diet, and Blue Seal Natural. Homemade diets that do not contain excess copper should include meats, poultry, fish, and dairy products. Foods with excessive copper should be excluded from the diet. These include eggs, liver, shellfish, organ meats, beans/legumes, mushrooms, chocolate, nuts, and cereals. Mineral supplements containing copper should also be avoided. Other treatment measures are supportive and symptomatic. These are discussed in detail in the section on management of hepatic disease.
Infectious Inflammatory Diseases
Primary infections involving the liver are rare causes of hepatitis in dogs and cats. It is not uncommon, however, to culture bacteria as a secondary event in noninfectious hepatic diseases due to decreased hepatic reticuloendothelial cell function. In addition to bacterial pathogens, parasitic and fungal infections can involve the liver.
Bacterial Hepatobiliary Infections
Because the liver plays a central role in processing portal products, receives an extensive arterial blood supply, has an important reticuloendothelial cell function, and has a direct connection to the intestine via the biliary tract, it is subject to infection by several routes, including hematogenous (portal or arterial) and ascending via the biliary system. However, bacterial infection of the liver only occurs under unusual circumstances, including patients receiving immunosuppressive drugs, and patients with hyperadrenocorticism, diabetes mellitus, severe enteritis, biliary stasis, septicemia, decreased hepatic blood supply, and devitalization or necrosis of hepatic tissue. When the source of infection originates from the bowel, treatment is aimed at bacteria normally found in the gut. In one study in cats with suppurative cholangiohep- atitis, bacteria cultured from hepatic biopsy specimens (in descending order of prevalence) were E. coli, Staphylococcus, α-hemolytic streptococcus, Bacillus, Actinomyces, Bacteroides, Enterococcus, Enter- obacter, and Clostridia. In another study of 14 dogs with hepatic abscesses, E. coli, Clostridium sp., Klebsiella pneumoniae, Enterococcus sp., Staphylococcus epidermidis, and Staphylococcus intermedius were the most common bacteria isolated. Many patients have polymicrobial infections.
Clinical Findings
Bacterial cholangiohepatitis or hepatic abscesses usually cause persistent fever and anorexia. Hepatomegaly is variable, as is abdominal pain or other signs of peritonitis. There may be other signs of systemic infection, including lymphadenopathy, or signs of infection of other organs such as pneumonia, endocarditis, urinary tract involvement, or enteritis. Other risk factors include biliary obstruction, inflammatory bowel disease, and pancreatitis.
Laboratory findings usually include increased activities of SALT and SAST. If there is involvement of the biliary system, there will be increased activities of serum ALP and GGT and possibly increased bilirubin concentration. There may be a neutrophilic leukocytosis and hyperglobulinemia (especially with chronic infection). Coagulopathies may be present.
Radiographic findings are usually normal; however, radiolucent areas within the liver or biliary system and/or gallbladder may be seen secondary to gas-forming bacteria. Radiopaque choleliths may also be seen. Ultrasound imaging may detect discrete abscesses if present, seen as multiple hypoechoic, hyperechoic, heterogenous, or anechoic areas within the liver. In cases of cholecystitis, ultrasonographic findings include a thickened gallbladder wall with sludge within the lumen.
A definitive diagnosis is made by hepatic biopsy. If discrete abscesses are suspected from radiographic or ultrasonographic findings, blind percutaneous biopsy methods are contraindicated. Aerobic and anaerobic cultures of the liver should be obtained whenever hepatic biopsy is performed. False-negative cultures can be obtained with prior antibiotic treatment.
Treatment involves an appropriate antibiotic based on hepatic biopsy specimen culture. If culture results are negative despite gross and histopathologic findings suggestive of bacterial infection, the antibiotic choice depends on the suspected route of infection. If the GI tract is the origin, antibiotic use should include coverage against anaerobes and gram-negative organisms. Good combinations include a fluoroquinolone and ampicillin or metronidazole. Fluoroquinolones have excellent gram-negative activity and are effective orally administered drugs for treatment of hepatobiliary bacterial infections. Ampicillin and metronidazole are indicated for anaerobic infections. Single-agent amoxicillin-clavulanic acid (Clavamox) is also a good choice due to this agent's broad spectrum of activity. Clindamycin is also very active against anaerobes and enters the liver in high concentrations. However, it should not be used with bile duct obstruction or severe impairment of hepatic function. Antibiotics must be administered for several months. If discrete abscesses are present, ultrasound imaging can guide an end point for therapy. Antibiotics are given for at least 1 month following resolution of ultrasound lesions.
If discrete abscesses are present, surgical intervention for drainage or resection may be necessary. Likewise, if severe cholecystitis is present, cholecystectomy may be indicated. Concurrent medical treatment with appropriate antibiotics is also indicated. In one study of 14 cases there was a high survival rate in those dogs that underwent surgical treatment for hepatic abscesses.
Cholestasis has also been associated with extrahepatic bacterial infections in dogs and cats. Typical findings include hyperbilirubinemia with variable increases in serum hepatic enzyme activities. Histologically there is bile pigment accumulation in hepatocytes with variable inflammatory changes.
Parasitic Hepatobiliary Infections
Parasitic infections of the liver and biliary tract are rare but are more common in certain geographic areas such as the southeastern United States. Cats are most frequently infected with trematode parasites because of their propensity to ingest intermediate hosts (land snails, fish, reptiles, and amphibians). Parasites reported to infect the liver and biliary system include Platynosomum, Heterobilharzia, Cytauxzoon, Opisthorchis, Amphimerus, Metorchis, and Clonorchis.
Clinical signs of fluke infestation are usually associated with extrahepatic biliary obstruction. Many cats with fluke infestation are asymptomatic; thus the detection of the presence of flukes may not mean that they are the causative agent for the observed signs. The diagnosis is established by detecting eggs in the feces or by histologic examination of hepatic biopsy tissue. The drug of choice for treatment of flukes is praziquantel (Droncit) at a dosage of 2.3 mg/lb body weight two times a day for 3 days. Nitroscanate (Lopatol) at a single dose of 45 mg/lb may also be effective. Some cats require corticosteroids for treatment of concurrent eosinophilic cholangiohepatitis.
Fungal Infections of the Liver
Systemic mycoses, including histoplasmosis, blastomycosis, and coccidioidomycosis, can affect the liver. Hepatic involvement usually reflects systemic involvement and is one of many tissues affected. The significance of hepatic involvement is that it often represents an easy means of detection of the offending organism. Hepatic enzyme elevations suggest hepatic involvement and may justify hepatic biopsy. This is often the least invasive means of establishing a definitive diagnosis. Histopathologic examination reveals a granulomatous hepatitis with the offending organism usually seen with special stains. Fungal culture of hepatic tissue is usually not necessary but may be helpful.
Noninflammatory Diseases of the Liver
Important noninflammatory diseases of the liver include vascular anomalies (portosystemic shunts, portal vein hypoplasia), metabolic derangements (feline hepatic lipidosis, steroid hepatopathy, glycogen-storage diseases), hepatic amyloidosis, urea cycle enzyme deficiencies, abnormal iron storage, and hepatic neoplasia. This discussion will review the more common of these disease entities.
Portosystemic Shunts
Portosystemic shunts (portal vascular anomalies, portacaval shunts, portosystemic vascular anastomoses) can occur as a congenital anomaly or as an acquired entity secondary to chronic portal hypertension. In the latter circumstance, elevated portal blood pressure leads to opening of fetal blood vessels that shunted portal blood away from the liver into the systemic circulation. These vessels normally close at birth. They act as a reservoir to handle the increased pressure load. Unlike congenital portosystemic shunts, these shunts are usually multiple, extremely tortuous, and variable in their extrahepatic location. The most common cause for acquired portosystemic shunting is cirrhosis. The remainder of this discussion will concern congenital portosystemic shunts.
Etiology of Congenital Portosystemic Shunts
Normally blood leaving the stomach, intestines, spleen, and pancreas enters the portal vein and flows through the liver before entering the hepatic vein and the systemic venous circulation. Portal blood thus contains absorbed nutrients, intestinal hormones (which are tropic to the liver), and bacterial products and toxins. In the fetus the function of the liver for processing these products is minimal, and thus there are vessels to shunt blood away from the liver into the systemic venous circulation. Normally these vessels close shortly after birth, allowing the establishment of hepatic circulation. When any of these vessels remain patent after birth, portosystemic shunting occurs. The cause of these anomalies is unknown.When blood is diverted away from the liver, hepatic atrophy results because of the lack of tropic factors present in portal blood (especially insulin and glucagon).
There are several types of congenital portosystemic shunts seen in dogs and cats. These include (but are not limited to) the following:
1. Patent ductus venosus with or without a hypoplastic portal system
2. Major solitary portal-caudal vena caval anastomosis
3. Portal vein atresia, associated with the development of multiple portal-caudal vena caval anastomoses
4. Major solitary portal-azygous shunt
5. Portal-azygous shunt with discontinuation of the prerenal segment of the caudal vena cava
6. Left gastric vein to caudal vena cava
7. The development of intrahepatic arterioportal fistula.
Approximately one fourth of congenital portosystemic shunts are intrahepatic in both dogs and cats, with most associated with a patent ductus venosus. Single extrahepatic shunts, with a major solitary portal-caudal vena caval shunt being the most common, constitute 50% of portosystemic shunts. Most intrahepatic shunts are found in large-breed dogs (Doberman pinscher, golden retriever, Labrador retriever, Irish setter, Samoyed, and Irish wolfhound), whereas most extrahepatic shunts are seen in small-breed dogs (miniature schnauzer,Yorkshire terrier, miniature poodle, and dachshund). Dogs with intrahepatic shunts may develop clinical signs at an earlier age than dogs with extrahepatic shunts, possibly due to a larger volume of blood flow through intrahepatic shunts. Approximately 2% of portosystemic shunts seen in small animals occur in cats.
The severity of clinical signs depends on the volume and location of the shunt. Clinical signs result from impairment of hepatic function, leading to hepatic encephalopathy. The most important factors that lead to encephalopathy are the accumulation of blood ammonia and other gut- associated encephalopathic toxins that are normally metabolized and cleared by the liver. With portosystemic shunting, ammonia and other toxins increase in the blood, leading to encephalopathy. In addition, increased benzodiazepine-like substances and amino acid derangements (increased aromatic amino acids) occur with portosystemic shunts, contributing to encephalo- pathic signs.
Clinical Signs
Most cases are diagnosed in patients less than 1 year of age; however, there have been cases in patients as old as 10 years at the time of diagnosis. There is no sex predilection. Clinical signs in patients with portosystemic shunts are highly variable. Because of the diverse nature of signs, the clinician must maintain a high index of suspicion in any young patient with unexplained signs compatible with a shunt to avoid missing the diagnosis. Clinical signs often change throughout the day or week. There may be an exacerbation of signs after feeding, reflecting ammonia generation from dietary substrates; however, this finding seldom occurs in most cases.
The most common clinical signs are chronic depression, retarded growth, and weight loss. Most dogs are stunted in appearance and “poor doers.” Additional clinical signs include chronic GI signs (vomiting, diarrhea), anorexia, polydipsia/polyuria, and neurologic signs. It is often the neurologic signs that are most suggestive of a shunt, and approximately 90% of cases have some degree of neurologic dysfunction related to hepatic encephalopathy. Neurologic signs are often variable and wax and wane over time, including depression, incoordination, behavioral changes (often aggressive), amaurotic blindness, seizures, dementia, and stupor. An additional abnormality is intolerance of anesthetic agents (often seen during routine neutering in the first year of life). Any young patient that has persistence of any of these signs (especially neurologic or behavior changes) should be suspected of having a portosystemic shunt. In the cat, ptyalism is a common sign, as are central nervous system (CNS) and GI signs. Any young cat with ptyalism and/or CNS signs should be evaluated for the presence of a portosystemic shunt.
Laboratory Findings
Routine hemograms and serum chemistry profiles may be normal. Often there will be a mild nonre- generative microcytic anemia, and target cells may be seen. Most patients have conspicuously normal (or mildly increased) serum hepatic enzyme activities despite hepatic failure. This is because the principal lesion is one of atrophy and lack of portal blood supply. Cholestasis and hepatocellular necrosis and leakage are not features of this disease. Approximately 50% of patients will have mild hypoalbuminemia (reflecting decreased albumin synthesis or increased volume of albumin distribution if ascites is present), and many patients (70%) have decreased BUN concentrations (reflecting decreased synthesis from ammonia).Virtually all patients have normal serum bilirubin concentrations. A fasting hypoglycemia may be seen.
Because many of these changes are nonspecific, hepatic function tests must be performed to document hepatic failure. Serum bile acid measurements and the ammonia tolerance test are the most sensitive tests to detect the presence of a portosystemic shunt, being abnormal virtually 100% of the time.
Approximately one third to half of dogs with portosystemic shunts have ammonium biurate crystals in the urine. These form because of the increased concentration of uric acid in the urine as a result of decreased hepatic conversion to allantoin and increased urine concentration of ammonia associated with hyperammonemia. When the urine is acidic and supersaturated with these substrates, crystallization and precipitation can occur. In addition to crystals in the urine, calculi can form in the kidney or less commonly in the bladder. These stones usually are composed of ammonium acid urate or uric acid. Often the presence of renal calculi is an important clue that a portosystemic shunt is present in a young patient. If calculi are removed and crystallographic analysis identifies that uric acid stones are present, the patient should be evaluated for the presence of a portosystemic shunt.
Radiographic Findings
The most reliable methods of confirming the presence of a portosystemic shunt are contrast radiography and nuclear scintigraphy. Plain radiographic findings are usually indicative of microhepatica, best determined by upright angulation of the stomach on a lateral projection. Because puppies and kittens normally have large livers relative to their body size, this finding should increase the index of suspicion of a portosystemic shunt. The diagnosis can then be confirmed by evaluating hepatic blood flow with contrast radiography or nuclear scintigraphy.
There are several techniques to evaluate hepatic blood flow, including intraoperative mesenteric portography, percutaneous splenoportography, and cranial mesenteric arterial portography. Intraoperative mesenteric portography is the most practical technique to be applied in general practice. In addition to identifying the shunt, mesenteric portography can also assess the residual portovenous flow into the liver for prognostic importance. Following an injection into a mesenteric or jejunal vein, radiographic contrast medium normally flows into the portal vein and arborizes into the liver. In patients with a portosystemic shunt, contrast medium will bypass the liver and be seen in the caudal vena cava or azygous vein. If the caudal extent of the shunt is cranial to T13, it is probably an intrahepatic shunt, whereas if the caudal extent of the shunt is caudal to T13, it is probably an extrahepatic shunt. Once a shunt is identified on the angiographic study, one can proceed with surgical correction during the same surgical procedure. Alternatively, the patient can be allowed to recover from the anesthetic procedure used to obtain the angiogram and a second procedure is then performed later (2 to 4 weeks or longer) to correct the shunt. I prefer the latter approach in small patients that are under anesthesia for a long time for the angiogram because of the potential for hypothermia and prolonged anesthetic recovery.
Portosystemic shunts can also be identified with ultrasonographic guidance or nuclear scintigraphy (see section on nuclear scintigraphy to evaluate the liver). If the latter diagnostic method is used, mesenteric portography may still be necessary to locate the shunt if it is not readily seen during exploratory laparotomy.
Pathologic Findings
Histopathologic findings in patients with portosystemic shunts include diffuse hepatic atrophy, lobular collapse, and proliferation of small hepatic arterioles. Atrophy is characterized by close proximity of the portal triads, compressed hepatic cords, and inconspicuous portal veins. These findings are usually diagnostic of a congenital vascular anomaly, and hepatic biopsy specimen analysis represents another method (in addition to radiographic findings) to obtain a diagnosis. However, these changes may be difficult to distinguish from portal vein hypoplasia without a macroscopic shunt (formerly known as hepatic microvascular dysplasia; see section starting on p. 334). The latter disorder has clinical and histopathologic features of a portosystemic shunt but has normal findings on mesenteric portography and/or nuclear scintigraphy. Degenerative changes in the brain suggestive of hepatic encephalopathy include leukopolymicrocavitation at the gray-white matter junction, spongiform degeneration, and cortical necrosis.
Surgical Treatment
The treatment of choice for single portosystemic shunts is surgical ligation, because long-term medical management is palliative rather than curative. Before surgical intervention, medical management may be necessary to stabilize the patient. Emergency treatment of hepatic encephalopathy includes cleansing enemas, oral antibiotics or lactulose administration (see section on management of acute hepatic failure). Surgical correction of multiple portosystemic shunts is usually not successful because underlying portal hypertension and hepatic pathologic abnormalities persist. (See the References or surgical textbooks for detailed descriptions of surgical techniques.)
As a general rule small-breed dogs have extrahepatic shunts and large-breed dogs have intrahe- patic shunts. Single extrahepatic shunts are usually readily identified at surgery and can thus be isolated and attenuated or occluded (depending on portal pressure). Surgical manipulation of intra- hepatic shunts is much more difficult, whereas extrahepatic shunt ligation is more adaptable to general practice. Most extrahepatic shunts are found terminating in the caudal vena cava between the left phrenicoabdominal and renal veins.
Once the shunt vessel is isolated, correction can be made by gradual occlusion using an ameroid constrictor (see below) or by occlusion with cellophane banding on suture material. If suture is used, portal pressures must be measured to determine whether complete occlusion is possible. This is readily done by placing a 3½ or 5 Fr feeding tube into a mesenteric vein and threading it into the portal vein. This is then connected to a saline manometer to measure pressure, with the zero level standardized at the level of the femoral triangle or heart. Normal portal pressure is 10 to 15 cm H2O (8 to 12 mm Hg), and most patients have normal or decreased portal pressure before shunt manipulation. If a shunt is completely ligated, fatal acute portal hypertension can develop. This results from splanchnic congestion and stasis of blood, with the rapid development of endotoxic shock. During shunt attenuation with a silk suture, portal pressure should not exceed 20 to 23 cm ^O (18 to 21 mm Hg) or 11 cm H2O (8 mm Hg) above baseline. The silk ligature is placed as close to the vena cava as possible and gradually tightened while measuring portal pressure. Observing splanchnic viscera for signs of stasis, including blanching of the bowel, hypermotility of the small bowel, and distended and pulsating jejunal arteries is also important. Monitoring central venous pressure may also be helpful to predict the presence of portal hypertension. With increased portal resistance and decreased portal venous flow, central venous pressure drops because of decreased venous return and splanchnic venous pooling.
To avoid the need to measure portal venous pressure and still allow complete occlusion of the shunt, attenuation can be accomplished with an ameroid constrictor.[†††††] This is a device that is shaped like a miniature donut that has a small opening allowing it to be placed around the shunt. It can be subsequently locked to prevent its removal after placement around the shunt vessel. The constrictors come in various inner diameters (3.5, 5.0, or 6.0 mm) so that they can be used on various-sized shunt vessels. The constrictor is made of hygroscopic casein material that is porous, surrounded by a metal outer ring. As the porous material is gradually saturated with peritoneal fluid, it expands. Because the outer metal ring prevents outward expansion, there is inward expansion, gradually occluding the central hole, which has the shunt vessel in it. In this manner the shunt is gradually occluded, usually over a period of 4 to 8 weeks. Because there is gradual occlusion, there is no need to measure portal pressures. As the shunt is occluded the hepatic vasculature becomes more perfused, thus acting to prevent portal hypertension. In general there is complete occlusion of single extrahepatic shunts using the ameroid constrictor in approximately 80% to 90% of cases.
Manipulation of intrahepatic shunts is technically more difficult. Several techniques have been described for attenuation of intrahepatic shunts. See the References and surgical literature for details. If possible, the shunt can be isolated before its entry into the liver or as it leaves the liver before entering the caudal vena cava. Otherwise the shunt is looked for by incising the prehepatic vena cava after occluding hepatic and vena caval blood flow and located by noting the abnormal irregular margins of the shunt vessel as it enters the vena cava from the inside. Alternatively, the shunt can be located by a transportal approach following vascular occlusion. If the shunt cannot be occluded completely without causing portal hypertension, a novel approach has been described that involves complete intrahepatic shunt closure along with the surgical creation of a portacaval shunt using an external jugular vein graft. An ameroid constrictor is placed around the graft to permit its gradual closure.
Dogs that have mild to moderate portal hypertension (20 to 23 cm H2O) following shunt manipulation usually have normal portal pressure within several weeks of surgery. These dogs often have ascites secondary to the transient portal hypertension, which disappears in 1 to 3 weeks as portal pressure drops. Often the silk ligature used to partially occlude a shunt will cause a reaction that results in gradual complete occlusion of the shunt over time.
A biopsy of the liver is always performed for histopathologic evaluation and occasionally for bacterial culture. If there are cystic or renal calculi concurrent with the portosystemic shunt, they can be removed during the procedure for shunt correction if the patient is stable and the anesthetic time is not excessive. Otherwise a second surgery is performed several weeks later to remove the urinary calculi. A follow-up mesenteric portogram and hepatic biopsy can be performed at this time to evaluate the shunt correction.
Postoperative monitoring is important to detect signs of severe portal hypertension, including abdominal pain, hemorrhagic diarrhea, and endotoxic shock leading to death. Fortunately, the use of an ameroid constrictor or intraoperative monitoring of portal pressure makes this an unusual complication. Hypoglycemia may occur if the patient is not eating, necessitating intravenous glucose supplementation. Intravenous crystalloids or colloids (if there is significant hypoalbumine- mia) are essential in the immediate postoperative period. Status epilepticus and generalized motor seizures can occur following shunt attenuation. These are usually first observed 3 days postopera- tively. The etiology of the seizures is unknown, but one theory is that there may be stimulation of brain receptors for benzodiazepines associated with the presence of the shunt. Following ligation of the shunt, seizures may result from withdrawal of benzodiazepine-like substances (documented to be present in portal blood of dogs with portosystemic shunts) following shunt ligation. The prognosis is poor in my experience despite control of status epilepticus. The role of prophylactic anticonvulsant drugs such as potassium bromide needs to be defined.
Medical management of chronic hepatic disease should continue as needed to manage signs of encephalopathy; however, most patients are asymptomatic shortly after surgical shunt correction. Patients should be evaluated 1, 3, and 6 months after surgery with serum biochemical and hepatic function tests (e.g., bile acids assay). Persistent abnormal function suggests incomplete shunt closure, concurrent portal vein hypoplasia, or the development of multiple extrahepatic shunts if portal hypertension results. If an ameroid constrictor or partial ligation is used to attenuate the shunt, nuclear scintigraphy (or mesenteric portography) is performed 2 to 3 months after surgery to evaluate for complete closure. If there is evidence ofincomplete shunt closure, nuclear scintigraphy is repeated 4 to 5 months following surgery. If there is still evidence of incomplete closure and the patient is still symptomatic, a second surgery could be performed for complete shunt closure. In one study 50% of dogs undergoing partial shunt closure developed complications associated with continued or renewed portosystemic shunting in a 4-year follow-up period despite excellent shortterm results. This suggests that a second surgery should be considered in these dogs.
Prognosis
The prognosis for medical management of congenital portosystemic shunts is poor. Most patients have progressive hepatic atrophy, and eventually signs of hepatic encephalopathy become refractory to medical management. Occasionally a patient will live to an old age (with or without medical therapy), although later in life such patients often have urate urinary calculi or signs of hepatic encephalopathy. These cases are uncommon, however.
The prognosis for single extrahepatic shunts with surgical correction is excellent, unless severe portal hypertension persists (which is unusual). Clinical improvement is often seen shortly after surgical correction. Hepatic biopsy specimen analysis obtained several months after surgical correction may be normal if there is no concurrent portal vein hypoplasia. The results of surgical ligation of extrahepatic portosystemic shunts in cats seem to be worse than in dogs, with only approximately 50% to 60% of cats having a favorable outcome.
The prognosis for intrahepatic shunts is more guarded due to the technical difficulties of the surgical correction and inability to completely attenuate the shunt without developing portal hypertension. Success depends in large part on the skill and experience of the surgeon.
Portal Vein Hypoplasia Without a Macroscopic Shunt (Formerly Hepatic Microvascular Dysplasia)
Portal vein hypoplasia without a macroscopic shunt (formerly referred to as HMD) refers to a microscopic pathologic malformation of the hepatic microvasculature. It is characterized by small intrahepatic portal vessels, portal endothelial hyperplasia, portal vein dilation, random juvenile intralobular blood vessels, and central venous mural hypertrophy and fibrosis. It is thought that these lesions allow abnormal communication between portal and systemic circulation. It is important to note that portal vein hypoplasia can occur as an isolated disease or in conjunction with macroscopic portosystemic shunts. In one large study 58% of dogs and 87% of cats with portal vein hypoplasia also had concurrent congenital portosystemic shunts. Dogs and cats with portal vein hypoplasia can have clinical signs similar to those of portosystemic shunts, including neurologic and GI abnormalities, as well as urate urolithiasis. Portal hypertension does not usually develop in dogs and cats with portal vein hypoplasia.
Breeds of dogs affected with portal vein hypoplasia are similar to those with congenital portosystemic shunts, including Yorkshire and Cairn terriers (a hereditary mechanism has been described in this breed). Reports of dogs with portal vein hypoplasia suggest that clinical signs and clinicopathologic features are similar to those of portosystemic shunts, although often not as severe. A recent study of 42 cases comparing dogs with portal vein hypoplasia alone and dogs with portal vein hypoplasia concurrent with portosystemic shunts revealed that dogs with portal vein hypoplasia alone were older and had higher values for mean corpuscular volume (MCV) and serum total protein, albumin, creatinine, cholesterol, BUN, and blood glucose concentrations. In addition, dogs with portal vein hypoplasia alone had lower preprandial and postprandial bile acid concentrations. The most discriminating variables for the two groups were postprandial bile acid concentrations, MCV, and serum albumin and cholesterol concentrations. However, there is a large overlap in values, suggesting that patients with presenting clinical findings of a congenital vascular anomaly must undergo an imaging study (nuclear scintigraphy or mesenteric portography) to detect patients with macroscopic portosystemic shunts, because these are amenable to surgical therapy. Further definition of portal vein hypoplasia requires hepatic biopsy. A surgical wedge biopsy or laparoscopic “spoon” biopsy is preferred because they provide more hepatic lobules for evaluation.
Treatment for portal vein hypoplasia is supportive because there is no macroscopic shunt to attenuate. Dietary measures and agents used to treat hepatic encephalopathy are described in the section on management of chronic hepatic disease. Many dogs remain asymptomatic with dietary therapy alone, although the prognosis is variable.
Steroid Hepatopathy
Steroid hepatopathy can result from excessive endogenous (hyperadrenocorticism) or exogenous (iatrogenic administration) amounts of glucocorticoids. It represents one of the most common causes of increased serum hepatic enzyme activities and the most common diagnosis on hepatic biopsy specimen analysis in dogs. Steroid hepatopathy occurs only rarely in the cat. The etiology of changes in the liver induced by glucocorticoids is unknown. The likelihood that an individual patient will develop steroid hepatopathy following glucocorticoid administration is variable and depends on individual sensitivity, type, route, and duration of administration of the glucocorticoid. Some patients show minimal changes in serum hepatic enzyme activities and morphologic changes in the liver even after chronic glucocorticoid administration, whereas other patients have increased serum hepatic enzyme activities and morphologic changes that persist for weeks after a single dose of a glucocorticoid. Changes can persist for several months after a single injection of a long- acting glucocorticoid or after chronic administration of oral glucocorticoids. Changes can also occur after topical or ocular administration of glucocorticoids.
Clinical Findings
Clinical signs of steroid hepatopathy range from asymptomatic to those associated with glucocorticoid excess. These signs include polyuria, polydipsia, polyphagia, endocrine alopecia, distended abdomen, and lethargy. There are usually no signs specifically related to hepatic failure with the exception of lethargy in severe cases. Hepatomegaly is often identified on abdominal palpation and on abdominal radiographs.
Laboratory Findings
There are usually mild to moderate increases in SALT and SAST activities, and marked increases in serum ALP and GGT activities in dogs with steroid hepatopathy. These increases are variable. Occasionally the magnitude of elevation in serum activities of the transaminases (ALT and AST) exceeds the magnitude of elevations of serum ALP and GGT activities. Serum albumin and bilirubin concentrations are usually normal (when these are abnormal, other causes should be looked for). Often the laboratory abnormalities seen with primary nonhyperbilirubinemic hepatobiliary disease and with steroid hepatopathy are similar. The presence of increased serum bilirubin concentration virtually eliminates steroid hepatopathy from the differential diagnosis of primary hepatobiliary disease.
The increase in serum ALP activity is attributed to an isoenzyme that is different from that induced by biliary stasis. This isoenzyme, also produced in the liver, is referred to as the steroid- induced isoenzyme of ALP. Laboratory methods are available to distinguish the steroid-induced isoenzyme from that induced by biliary stasis. However, the steroid-induced isoenzyme of ALP is variably elevated with many primary hepatobiliary diseases and therefore measurement of its activity is not a useful test to determine the presence of steroid hepatopathy. Though there is not a specific isoenzyme of GGT induced by glucocorticoids, it is important to note that the magnitude of increased serum GGT activity induced by glucocorticoids often parallels that of ALP and that serum GGT activity cannot be used to distinguish steroid hepatopathy from primary hepatobiliary disease.
Results of hepatic function tests, including serum bile acids and blood ammonia concentrations, are variable with steroid hepatopathy. Serum bile acids can be normal or slightly to moderately elevated (less than 75 to 100 μmol∕L). Marked increases in serum bile acids are unlikely to result from steroid hepatopathy, and other hepatobiliary diseases should be considered. Blood ammonia concentration and the ammonia tolerance test results are usually normal with steroid hepatopathy.
Histopathologic Findings
Histopathologic abnormalities are usually highly suggestive for steroid hepatopathy. There is usually marked vacuolization and ballooning of hepatocytes in a centrilobular or diffuse distribution. The vacuoles are thought to be due to glycogen deposition. There might also be a variable degree of hepatic necrosis.
Diagnosis
If laboratory and clinical signs are compatible with steroid hepatopathy or the patient is asymptomatic, steroid hepatopathy should be ruled out before hepatic biopsy. This can be done with a history of glucocorticoid administration or appropriate laboratory tests such as the adrenocorticotropic hormone (ACTH) stimulation or low-dose dexamethasone suppression tests. If the diagnosis is still uncertain, hepatic imaging with ultrasound (usually revealing hyperechoic and sometimes mottled parenchyma) and biopsy are warranted. Biopsy specimen analysis usually readily distinguishes steroid hepatopathy from other hepatic diseases. Hepatic biopsy specimen analysis is another method of diagnosing hyper- adrenocorticism when other laboratory tests are inconclusive.
Treatment
Treatment includes elimination of the source of excess glucocorticoids. If the source is exogenous administration, corticosteroids should be discontinued if their administration is not necessary or decreased by at least 50% if their ongoing use is considered important. If clinical signs such as lethargy are present and persist, alternative immune- modulating drugs may be substituted if appropriate (such as azathioprine, cyclophosphamide, or cyclosporine). Hyperadrenocorticism should be treated if clinically indicated. The laboratory and morphologic changes in the liver seen with steroid hepatopathy are completely reversible when the source of excess glucocorticoids is removed.
Feline Hepatic Lipidosis Syndrome
Feline hepatic lipidosis is a common condition in cats that results in intrahepatocyte accumulation of triglycerides because of abnormalities in lipid metabolism and is associated with severe hepatic dysfunction. In the dog and cat, accumulation of fat in hepatocytes can be associated with a number of causes, including endocrine, nutritional, metabolic, and toxic abnormalities. These causes are listed in Box 9-13. In these instances there is a known underlying cause. In the syndrome referred to as feline hepatic lipidosis, there is no known underlying cause and the disease occurs as an idiopathic entity. This discussion will be confined to this syndrome.
BOX 9-13
Causes of Lipid Accumulation in the Liver
Nutritional
Prolonged overnutrition
Starvation
Obesity
Deficiency of essential nutrients (lipotrophic agents)
Protein deficiency
Endocrine disorders
Diabetes mellitus
Toxins
Drugs
Bacterial endotoxins
Chemicals
Plants
Hypoxia
Idiopathic
Feline hepatic lipidosis syndrome
Etiology
In some cases an underlying disease is associated with feline hepatic lipidosis, although the majority of cases are idiopathic. The most common underlying diseases identified are pancreatitis, inflammatory bowel disease, and cholangiohepatitis. The etiology of idiopathic feline hepatic lipidosis is unknown. Theories proposed are based on the liver's role in triglyceride metabolism. Triglycerides accumulate in the liver when the rate of hepatic uptake or synthesis exceeds the rate of removal. Sources of triglycerides include de novo synthesis in the liver and from fatty acids in the systemic circulation (derived from dietary sources and from adipose stores). Once in the hepatocyte, fatty acids are esterified to triglycerides and phospholipids or oxidized within the liver. Triglycerides are released from hepatocytes primarily as very-low-density lipoproteins (VLDLs).
When the ability of the liver to excrete or oxidize triglycerides is exceeded by triglyceride supply, triglycerides will accumulate in the hepatocyte. Therefore hepatic lipid accumulation can result from (1) excessive hepatic triglyceride synthesis, (2) impaired fatty acid oxidation within the hepatocyte, (3) impaired fatty acid transport from the liver as VLDL, and (4) changes in the nutritional or hormonal status or toxic influences on hepatocellular functions that adversely affect triglyceride metabolism. In addition to the possibility that excessive triglyceride storage is damaging to the hepatocyte, excessive hepatic triglyceride accumulation may indicate an abnormality in cellular metabolism that may also affect other cell functions.
It is not known how these mechanisms are involved in the pathogenesis of feline hepatic lipidosis. Anorexia in previously obese cats is an important feature of this disorder and may initiate fatty deposition in the liver. During starvation, free fatty acids increase because of release from adipose tissue. Obesity probably predisposes to this process. There is excessive mobilization of fatty acids, which are subsequently incompletely metabolized in the liver, leading to accumulation within hepatocytes. Some cats have an underlying systemic disease that initiates the anorexia and is concurrent with hepatic lipidosis, although in most cases an underlying disorder is not identified. It has also been proposed that cats with hepatic lipidosis are diabetic or prediabetic, although this is speculative and there is little evidence for this. It is possible that an insulinresistant state would result in continued release of fatty acids from adipose tissue and subsequent accumulation in the liver.
It is not known what metabolic or hepatocellular derangements prevent triglyceride removal from the liver, although some metabolic and ultra- structural abnormalities have been identified. In ultrastructural studies, cats with hepatic lipidosis have hepatocellular organelles and nuclei displaced to the cell periphery, resulting in compression of the lumen of bile canaliculi. This contributes to cholestasis and bile acid retention. There is also a unique abnormality in that there is a relative paucity of peroxisomes (rather than the up-regulation expected in circumstances of increased fatty acid oxidation), organelles important in the preprocessing of long-chain fatty acids before their presentation for mitochondrial oxidation. Peroxisomes take on an increased role in circumstances of mitochondrial dysfunction. Unlike mitochondria (which are dependent on carnitine), peroxisomal fatty acid oxidation and transport is facilitated but not dependent on carnitine. Peroxisomes are also involved in bile acid synthesis, and subsequent peroxisome dysfunction has lead to abnormal bile acid profiles in cats with hepatic lipidosis. Thus these defects in peroxisomes and increased dependency on peroxisomal oxidation may increase “oxidative stress” on hepatocytes.
There is also speculation that there is a relative carnitine deficiency, resulting in hepatocellular triglyceride accumulation in cats with hepatic lipidosis. It has been shown that plasma, urine, and hepatic tissue carnitine concentrations are normal (or elevated) in cats with hepatic lipidosis. However, it is possible that there may be inadequate carnitine for the quantity and rate of mitochondrial oxidation and disposal of accumulating acetyl CoA. Furthermore there is increased urine concentration of short-chain acyl-carnitines in cats with hepatic lipidosis, reflecting an increased production or carnitine-facilitated removal from the liver. This could allow a route for elimination of excess fatty acids from the liver. Finally, there is evidence that carnitine supplementation to cats with hepatic lipidosis improves recovery rates.
Previous theories have been disputed as well. Orotic acid is a toxin capable of inducing hepatic lipidosis in other species by impaired phospholipid synthesis and VLDL transport from the liver. Arginine deficiency (seen in cats with hepatic lipidosis) can lead to orotic acid accumulation because arginine is essential for normal urea cycle function, and impaired urea cycle function is associated with production of orotic acid precursors. However, attempts to produce hepatic lipidosis in cats with orotic acid administration were unsuccessful, and urine orotic acid concentrations are normal in cats with hepatic lipidosis. Vitamin B12 deficiency has also been speculated to be associated with hepatic lipidosis. In one description of 96 cats with hepatic lipidosis, vitamin Bn deficiency was not documented. In that series, inspection of plasma and urine for unusual fatty acids reflecting site-specific impaired mitochondrial oxidation did not reveal unique moieties, suggesting no obvious defect in a particular mitochondrial enzyme.
Clinical Features
As mentioned earlier, most cats with hepatic lipidosis are obese. There is usually a period of anorexia followed by signs typical of hepatic failure. In some cases a known illness, stressful event (e.g., boarding, travel), or diet change may cause the initial period of anorexia. In most cases, however, no initiating cause is known. When hepatic lipidosis occurs, clinical signs include inappetence, weight loss, vomiting, and jaundice. Physical examination findings include obesity with evidence of dorsal muscle wasting, jaundice, and possible hepatomegaly.
Laboratory and Radiographic Findings
Laboratory findings include marked elevations in serum hepatic enzyme activities, especially ALP and to a lesser extent the transaminases (SALT, SAST).Because the half-life of ALP is very short in the cat (6 hours), activity of this enzyme is only increased in the serum with severe hepatobiliary disease. In hepatic lipidosis the activity of ALP is usually markedly elevated and often higher than in any other form of hepatic disease in cats. Hepatic lipidosis is also the most common hepatobiliary disease in the cat to result in a magnitude of increased ALP activity exceeding that of serum GGT activity. Serum total bilirubin concentration is usually increased and reflects the degree of intrahepatic cholestasis. Coagulation abnormalities (especially elevated PIVKA times) are also common, occurring in almost 50% of cats in one study. However, overt bleeding (including from hepatic biopsy sites) is rare in my experience. Hypokalemia was present in 25% to 30% of affected cats and was found to be a negative prognostic factor. Postprandial serum bile acid concentrations are almost always abnormally elevated. Hypophosphatemia was present in 10% to 15% of cats in one report. Oral alimentation may result in a further decline in serum phosphorus concentration, resulting in clinical manifestations such as hemolytic anemia. Radiographs usually reveal normal hepatic size or hepatomegaly.
Ultrasonographic Findings
Ultrasonographic findings are almost always abnormal in feline hepatic lipidosis. Findings include overall increased echogenicity of hepatic parenchyma compared with falciform fat. In one study this finding was seen in 100% of cats with hepatic lipidosis. However, other studies have also found this relationship in diseases other than hepatic lipidosis, making this a nonspecific finding. In addition, there is increased beam attenuation by the liver, and borders of hepatic vessels are difficult to visualize. Other underlying disorders, such as pancreatitis, may also be detected with an ultrasound examination.
Pathologic Findings
The diagnosis of feline idiopathic hepatic lipidosis is based on histologic findings and the absence of other concurrent diseases that are known to cause lipid accumulation in the liver (see Box 9-13). Typical histopathologic features are a diffuse lobular fatty infiltration within individual hepatocytes. The lipid accumulation is usually macrovesicular in nature, although it can be microvesicular in some cats. Usually there is evidence of intrahepatic cholestasis. The diagnosis can often be made by analysis of fine needle aspiration of the liver or impression smear made of hepatic biopsy specimens. Cytologic features include vacuolated hepatocytes with minimal inflammation. However, cytologic examination cannot exclude the presence of concurrent diseases such as cholangiohep- atitis and lymphoma.
Grossly the liver in cats with hepatic lipidosis is usually large, friable, and has slightly rounded margins with a smooth surface. The color is usually yellow with an accentuated lobular pattern.
Treatment
If precipitating causes of the anorexia can be identified, they should be addressed. These include environmental influences such as diet changes and boarding. If concurrent diseases are identified, such as cholangiohepatitis, inflammatory bowel disease, or pancreatitis, they should be treated.
The goal of treatment is to reverse the metabolic changes that resulted in mobilization of free fatty acids occurring during starvation. This is usually accomplished with aggressive forcefeeding. In mild cases, methods to stimulate voluntary oral intake may be effective. Heating food and adding seasoning and salt substitutes to food may be helpful. However, tube feeding seems to be necessary in most cases. This is best accomplished with a PEG tube (No. 18 to 20 Fr), esophagos- tomy tube (No. 18 to 20 Fr), or nasoesophageal tube (No. 5 Fr). My preference is to place a PEG tube in most cases, often when the cat is under general anesthesia for the hepatic biopsy procedure if analysis of an impression smear of the biopsy sample is suggestive of hepatic lipidosis. This avoids the stress of a second anesthetic procedure. However, there are some cats that are clearly not stable enough to undergo an anesthetic procedure for placement of a feeding tube. In these cases, cats often never recover completely from the procedure. In these patients it is usually much safer to obtain a biopsy specimen under local anesthesia or to rely on cytologic analysis of a fine needle aspirate. This is then followed by placement of a nasoesophageal tube with the cat awake. This allows administration of a liquid nutritional formula (such as CliniCare [Pet-Ag]) on a temporary basis. Constant administration with an infusion pump or gravity flow (versus bolus feeding) is also helpful if vomiting is a problem during the initial few days of treatment. These cats often have electrolyte disturbances such as hypokalemia and should also be stabilized with appropriate intravenous fluids (non—lactate-containing fluids supplemented with potassium chloride or potassium phosphate).When the cat is more stable, a PEG or esophagostomy tube is then placed for long-term use at home. These large-bore tubes are preferred because they are more comfortable to the cat than nasoesophageal tubes and allow the owner to feed blended cat food at home.
The total calorie intake should be 28 to 36 kcal/lb body weight per day. If a large-bore tube is used, a balanced commercial cat food gruel (e.g., Hills Feline p/d [674 kcal per can] or c/d [604 kcal per can]) can be used as the feeding solution. These are generally diluted 1:1 with water, to make a gruel containing approximately 0.75 kcal/ml. They can also be diluted 1:2 with water to make a gruel containing approximately 1.0 kcal/ml. Restricted protein diets are not indicated unless there are overt signs of hepatic encephalopathy present (such as ptyalism). Initially feeding is started at one half the calculated amount for the first 24 to 48 hours. The calculated daily requirement is divided into four to six feedings per day initially. Eventually most cats tolerate the necessary volume in three to four feedings per day. The volume should not exceed 14 ml/lb body weight at any feeding. Attempts are made to gradually increase the feeding volume to maintenance over the first 3 to 5 days.
Antiemetics may be necessary to prevent vomiting during the first few days (or longer) of treatment in many cases. The drug of choice is metoclopramide (Reglan). This is given at a dosage of 2.5 to 5 mg 30 minutes before feeding. In most cases it works when given through the feeding tube (a liquid form is available for this purpose). Occasionally subcutaneous administration or constant intravenous infusion may be necessary. Other strategies to control vomiting include the addition of other antiemetics such as prochlorperazine (Compazine), chlorpromazine, or ondansetron. Other strategies are to administer a liquid enteral formula (CliniCare) by constant infusion or to decrease the amount of water added to the cat food gruel to decrease the volume administered and still provide the same amount of calories.When diluted with one part water to two parts food, the gruel contains 1.0 kcal/ml.
Various dietary supplements have been proposed to be helpful in treating cats with idiopathic hepatic lipidosis. Carnitine supplementation seems to improve survival rates and shorten recovery times. In one report (n = 57) supplementation with L-carnitine (250 to 500 mg/day) was advocated based on the possibility that there is a relative carnitine deficiency. Compared with historical controls, cats that received L-carnitine had a recovery rate of 81% compared with a recovery rate of 37% in cats that did not receive L-carnitine. Cats that received L-carnitine and gastrostomy tube feedings had a recovery rate of 89% (cats that did not receive L- carnitine but had gastrostomy tube feedings had a recovery rate of 29%). In this report, taurine supplementation (250 to 500 mg/day) was also advocated because many cats with hepatic lipidosis have decreased serum taurine concentrations. Taurine is important because this amino acid is used for obligatory bile acid conjugation in the cat and may modify the injurious potential of retained bile acids and increase their renal excretion. Thiamine should also be provided (100 mg by injection or orally two times a day for 3 days) if there is evidence of thiamine deficiency (ventral neck flexion). Vitamin E may be helpful to minimize oxidative hepatic injury. SAMe administration (9 mg/lb/day) may also help speed recovery.Vitamin K supplementation is used if overt bleeding is detected or suspected. If there is prolonged recovery (rare), ursodeoxycholic acid may be beneficial to help stabilize peroxisomes and because cholestasis is usually present (see earlier section on use of ursodeoxycholic acid in cats with cholangiohepatitis). In most cases ursodeoxycholic acid is not necessary because most cats recover or die before this drug can alter the outcome or provide hepatoprotection. Supplementation with L-citrulline (to promote ure- agenesis and minimize synthesis of orotic acid) and choline (required for phospholipid synthesis in VLDL production) have also been empirically recommended.
Low-dose insulin therapy is indicated only if the cat is overtly diabetic. There is no evidence that arginine supplementation is helpful in this disease. Lipotropic agents containing methionine have no therapeutic benefit and are contraindicated because they can result in the production of encephalopathic toxins (mercaptans). Use of glucocorticoids and anabolic steroids should likewise be avoided.
Prognosis
The prognosis is fair to good in most cases. I have been successful in treating approximately 80% to 90% of cats, with the recovery rate dependent on how stable the cat is at the time of presentation, the aggressiveness of force-feeding, and the ability to control vomiting. Virtually all cats recover if they survive beyond the first few days. The earlier they are treated, the higher the recovery rate. Spontaneous recovery is rare. Force-feeding may need to be prolonged (3 to 12 weeks). Most owners are able to manage gastrostomy or esophagostomy tube feeding at home during this period. Once there is normalization of biochemical parameters, tube feeding is gradually decreased and the cat should be coaxed to eat on its own. Once the cat is eating completely on its own without any tube feedings for 1 to 2 weeks, the tube is pulled. There is no evidence that affected cats are prone to recurrences following initial recovery or that there is residual hepatic damage. To prevent hepatic lipidosis, nutritional support should be provided to obese cats that stop eating because of other diseases.
Diseases of the Gallbladder and Bile Duct
Disorders of the gallbladder and bile duct are uncommon in small animals. Clinical signs depend on whether there is obstruction to the flow of bile or infection of the extrahepatic biliary system.
Bile Duct Obstruction
Etiology
Causes of common bile duct obstruction are listed in Box 9-1. Because the extrahepatic bile duct goes through the pancreas before it enters the duodenum, disease in the area of the pancreas can result in obstruction. Though pancreatitis is the most common cause of bile duct obstruction, most cases of pancreatitis do not result in bile duct obstruction.
Clinical Findings
Many patients with bile duct obstruction are symptomatic for their underlying disease (for example, patients with pancreatitis often have vomiting and abdominal pain). Signs associated with bile duct obstruction include weight loss, jaundice, anorexia, acholic feces, and vomiting. If the cause of the obstruction is not inflammatory in nature, clinical signs are often surprisingly mild. Often jaundice is the first sign observed.
Laboratory findings include elevations in serum hepatic enzyme activities. Often the serum activities of the cholestatic enzymes (ALP and GGT) are disproportionately elevated compared with serum activities of the transaminases (ALT and AST). However, this is not a consistent finding and many cases of intrahepatic cholestasis have similar biochemical profiles. Serum total bilirubin concentration is also elevated to a variable degree depending on the degree of obstruction. The relative amounts of direct (conjugated) and indirect (unconjugated) bilirubin are variable and often indistinguishable from changes seen with intra- hepatic cholestasis. Therefore laboratory findings identify hepatobiliary disease but do not aid in distinguishing intrahepatic from posthepatic causes of cholestasis. This is currently best accomplished with ultrasonography. Ultrasonographic findings include common and intrahepatic bile duct distention, as well as gallbladder enlargement. These changes take several days to occur following obstruction. Dilation progresses from common bile duct to peripheral intrahepatic bile ducts over a period of 5 to 7 days following bile duct obstruction. Enlarged intra- hepatic bile ducts can be distinguished from portal vessels by their tortuosity, irregular branching pattern, and abrupt and variable changes in lumenal diameter. In addition, the cause of obstruction (such as neoplasia) is often identified on an ultrasonographic examination.
Hepatobiliary scintigraphy has also been used to distinguish extrahepatic biliary obstruction from intrahepatic disease. Following injection of 99mTc-diisopropyl iminodiacetic acid (di- sofenin) into patients with unobstructed biliary tracts, there is nuclear activity in the intestine within 3 hours. There is failure to visualize the intestine with nuclear imaging by 3 hours in patients with biliary obstruction. In one study, this method was 83% sensitive and 94% specific (91% accurate) in diagnosing extrahepatic biliary obstruction. Finally, hepatic biopsy often suggests the presence of extrahepatic biliary obstruction.
Treatment
Treatment of bile duct obstruction depends on the underlying etiology (see Box 9-1). In most cases, specific medical therapy is not possible and symptomatic care or surgery is necessary. Approximately 80% of patients with pancreatitis that results in bile duct obstruction will eventually resume normal bile flow without surgical intervention if given appropriate supportive care and enough time. In these cases obstruction is probably associated with acute edema and inflammation around the bile duct. When this resolves, bile duct patency returns. Therefore if pancreatitis is suspected as the cause of biliary obstruction, supportive care is warranted in the initial period. If there is no biochemical or clinical improvement within 2 weeks, patency is unlikely to spontaneously occur and surgical intervention is warranted. During this period, nutritional support with jejunostomy tube feeding or total parenteral nutrition (TPN) may be required. There is no specific medical therapy for bile duct obstruction caused by pancreatitis.
Surgical treatment is usually necessary for most other causes of bile duct obstruction. Surgery is intended to establish patency of the extrahepatic biliary system with the intestine. The procedure used depends on the location of the obstruction, the degree of distention of the common bile duct and gallbladder, the presence of concurrent cholecystitis, and the underlying disease. The most common procedures performed are cholecystoje- junostomy or cholecystoduodenostomy. If the gallbladder is infected and the common bile duct is large enough, a cholecystectomy and choledo- choenterostomy is indicated. A detailed description of these procedures is beyond the scope of this chapter. The reader is referred to the surgical literature for more information. Recurrent cholangitis and/or cholecystitis can be sequelae to biliary-enteric anastomoses; however, this is seldom a clinical problem if a large enough stoma is created. Clinical signs include depression, fever, and vomiting. Empirical antibiotic administration is usually effective in controlling these episodes.
Cholelithiasis and Choledocholithiasis
Etiology
Choleliths are rare in dogs and cats, and the etiology is unknown. Most theories implicate bile stasis, infection, and changes in bile composition. Choleliths in dogs and cats have been reported to contain primarily cholesterol and bilirubin. Relative percentages of these components are variable, and mixed stones often occur. Additional components include calcium, magnesium, and oxalates. If choleliths are analyzed by methods used for cystic calculi, cholesterol and bilirubin contents will not be determined.
Clinical Findings
Cholelithiasis is usually asymptomatic unless associated with cholecystitis or obstruction of bile flow. In one report approximately 75% of choleliths were discovered at necropsy and were not associated with clinical signs. When clinical signs are present, they are often intermittent.When cholecystitis is present, there is often fever, abdominal pain, and vomiting. When bile duct or cystic duct obstruction occurs, jaundice and other signs of extrahepatic biliary obstruction are seen. Physical examination findings usually reflect the degree of abdominal pain and jaundice.
Laboratory findings may be normal or similar to those seen with extrahepatic biliary obstruction, including increased serum activities of hepatic enzymes and bilirubin concentration.
Radiographic findings depend on whether choleliths are calcified. In most cases there is not enough calcium in the stones to make them radiopaque. However, when they are calcified, they are seen in the area of the gallbladder or rarely in the area of the common bile duct. Ultrasonography is the modality of choice in detecting choleliths, because the gallbladder is readily seen on an ultrasonographic examination. Choleliths appear as a hyperechoic area with a hypoechoic acoustic shadow. It can also be determined by ultrasonography whether there is gallbladder and biliary duct distention. A thickened gallbladder wall with inspissated bile suggests the presence of cholecystitis.
Treatment
Surgical intervention is usually the treatment of choice unless the patient is asymptomatic. Usually the procedure of choice is cholecystectomy, especially if there is concurrent cholecystitis. An alternative is cholecystotomy and stone removal. If there is common bile duct obstruction that cannot be relieved, a cholecystoenterostomy or choledo- choenterostomy will be necessary. At surgery bile should be cultured aerobically and anaerobically, so an appropriate antibiotic can be administered to manage concurrent cholangitis and cholecystitis. Additional therapeutic methods used to manage choleliths in humans include endoscopic removal, chemical dissolution, and extracorporeal shock wave lithotripsy. These methods have not been evaluated in small animals. A high-protein, low- cholesterol diet might be helpful to prevent recurrences.
Cholecystitis
Etiology
Cholecystitis is rare in the dog and cat. Predisposing factors include cholelithiasis, bile stasis, ascending biliary tract infection, and bacteremia with secondary cholangitis and cholecystitis. Cholecystitis is most commonly associated with complete or partial bile duct obstruction. Necrotizing cholecystitis often results in either chronic or acute gallbladder rupture with secondary bile peritonitis.
Clinical Findings
In mild cases, signs may be intermittent and include vomiting, fever, and abdominal pain. In acute necrotizing cholecystitis, signs include vomiting, anorexia, abdominal pain, and fever. Many of these patients will show signs of shock, including increased heart rate, pale mucous membranes, poor capillary refill, and weak pulses. When gallbladder rupture occurs, signs of bile and septic peritonitis will result.
Laboratory findings in severe cases include a neutrophilic leukocytosis with a left shift, hypoproteinemia, hypoglycemia, and increased BUN concentration. These changes are associated with sepsis and endotoxic shock. In addition, there are usually increased serum hepatic enzyme activities and increased serum bilirubin concentration if there is biliary obstruction. Abdominal paracentesis may reveal evidence of septic or bile peritonitis.
Radiographic findings include decreased abdominal detail if there is leakage of bile and peritonitis. If choleliths are present, they may be seen radiographically if they are calcified. Some cases have gas in the gallbladder (emphysematous cholecystitis) if there is a gas-forming organism involved (usually Clostridium spp. or E. colt). Ultrasonographic findings include gallbladder and biliary duct distention, cholelithiasis, thickening of the gallbladder wall, and inspissated bile.
Treatment
In mild cases treatment involves an appropriate antibiotic based on results of bile culture. In these patients the prognosis is fair. In severe cases, including those with necrotizing or emphysematous cholecystitis, the treatment of choice is cholecystectomy. In many cases the gallbladder is ruptured at the time of surgery. In this situation cholecystectomy and exploration of the abdomen for stones that escaped the gallbladder are required. Patency of the bile duct must be established and treated appropriately with diversion procedures if necessary. In cases without gallbladder rupture, it is still advisable to remove the gallbladder because it will be easier to treat the infection if the source is removed. Aerobic and anaerobic cultures of bile, calculi, and the gallbladder wall are mandatory to determine appropriate antimicrobial therapy. Pending culture results, a combination of an aminoglycoside or quinolone and ampicillin is recommended. In one large study the most common bacteria isolated were E. coli. Other bacteria cultured included Klebsiella sp., Clostridium sp., and Pseudomonas sp. Aggressive fluid support is also mandatory.
Prognosis
The prognosis is guarded to poor in severe cases. Early diagnosis and surgical intervention is the key for successful therapy. Death is usually attributed to sepsis, shock, peritonitis, and stress of anesthesia. Therefore patients that show signs compatible with cholecystitis should have the diagnosis aggressively pursued with serial abdominal paracentesis, ultrasonography, peritoneal lavage, abdominal radiography, and serial hemograms. When the diagnosis is suggested, surgical exploration should not be delayed.
Hepatic Neoplasia
Incidence
The liver is frequently affected with primary or metastatic neoplasia. Primary tumors account for 0.6% to 1.3% of all neoplasms in the dog. Metastatic tumors occur at least twice as frequently as primary tumors. Hepatic neoplasia occurs less frequently in the cat with the exception of malignant lymphoma and myeloproliferative diseases. The prevalence of hepatic neoplasia in the cat is 1.5% to 2.3%. The most common primary hepatic neoplasms in the dog in order of frequency are hepatoma, hepatocellular carcinoma, cholangiocarcinoma (bile duct carcinoma), fibroma, fibrosarcoma, hemangioma/heman- giosarcoma, leiomyoma, osteosarcoma, and hamartoma. The most common metastatic tumors in the dog that involve the liver are lymphoma and hemangiosarcoma. Other important primary sites are the mammary glands, adrenal gland, pancreas, bowel, bone, lung, and thyroid gland. Metastasis to the liver can occur via the portal vein, hepatic artery, lymphatics, or by direct extension. Spread from the portal circulation is most common. In the cat, malignant lymphoma and myeloproliferative diseases are the most common metastatic tumors. Nonhematopoietic hepatic neoplasms in cats include benign bile duct adenomas, bile duct adenocarcinomas, and hepatocellular carcinoma. Most hepatic tumors, with the exception of lymphoma, are seen in older patients. Hepatic neoplasia is reviewed in detail in Chapter 11.
Clinical Signs
Clinical signs of hepatic neoplasia depend on the extent of involvement. In many cases of primary neoplasia, especially hepatic adenomas and adenocarcinomas, signs are not seen until the tumor is very advanced. When symptomatic, patients show signs typical of other hepatic diseases, including anorexia, lethargy, vomiting, polyuria, and polydipsia. If there is involvement of the biliary system by direct involvement (cholangiocarcinoma) or by impingement of an extrahepatic bile duct, jaundice may be seen. In many cases hepatomegaly can be detected on physical examination. Clinical signs of metastatic neoplasia involving the liver tend to be more severe earlier in the disease, because more of the liver is usually affected. In addition to signs typical of hepatic failure, patients may also show signs typical of their primary tumor location.
Treatment and Prognosis
The treatment of hepatic neoplasia depends on the type of tumor and extent of involvement. Hepatomas and hepatocellular carcinomas grow very slowly and are often localized to a single lobe of the liver. They are often amenable to surgical resection and have good long-term prognosis. Up to 75% of the liver can be resected without significant hepatic dysfunction, and regeneration usually occurs within 6 to 8 weeks. Less commonly, hepatocellular carcinomas are nodular or diffuse. In these cases the prognosis is poor. Cholangiocarcinomas are usually widespread and either massive or diffuse. They are currently untreatable, usually rapidly progressive, and highly metastatic and have a grave prognosis.
Metastatic tumors are only treatable with chemotherapy. Lymphoma is the most responsive tumor to chemotherapy. Various protocols have been described, and most consist of various combinations of prednisone, cyclophosphamide (Cytoxan), vincristine (Oncovin), doxorubicin (Adriamycin), and L-asparaginase (Elspar). The reader is referred to veterinary oncology literature for details of these protocols. The prognosis with hepatic involvement is similar to that of multicentric involvement. In cats with well-differentiated lymphocytic lymphoma of the liver, the prognosis is better in my experience, with survival times usually between 1.5 and 2 years or longer when treated with a combination of prednisone and chlorambucil (Leukeran). Hemangiosarcoma can also be palliated with chemotherapy (using doxorubicin plus dacarbazine, or the combination of vincristine, doxorubicin, and cyclophosphamide). In humans hepatic arterial infusions of chemotherapeutic agents or embolization agents (such as iodinated poppyseed oil) using a pump delivery system or via angiography procedures are more efficacious than systemic administration for certain tumors, allowing higher regional drug concentrations. These methods have not been evaluated extensively in small animals.