Neonatal Encephalopathy
David Wong
NE is one of the most common neurologic diseases affecting neonatal foals with a reported incidence of 1% to 2% of all equine births.197 Other synonyms for NE reflect proposed etiologies of the disease, such as hypoxic-ischemic encephalopathy (HIE), perinatal asphyxia syndrome (PAS), and neonatal maladjustment syndrome; alternatively, more descriptive names reflecting the clinical manifestations of NE, such as barker, wanderer, or dummy foal, also have been used.6 It is important to note that foals with NE may have a hypoxic-ischemic event contributing to the disease process, but it is not uncommon
Seizure Disorders in the Neonatal Foal
Neonatal encephalopathy Bacterial meningitis Viral encephalitis
Benign juvenile epilepsy Lavender foal syndrome Kernicterus
Hepatic failure Congenital brain disease Hydrocephalus, hydranencephaly
Head trauma Hypoglycemia Electrolyte disorders
Hypocalcemia, hyponatremia, hypernatremia Tetanus
Toxicities
Ivermectin, moxidectin Glycogen storage disease IV Hyperammonemia of Morgan foals
for foals with NE to have no evidence of hypoxia-ischemia and an apparently normal birth.
Therefore other associated pathophysiologic mechanisms, such as excessive neurotransmitters, inflammation, and/or abnormal postpartum hormonal concentrations, also might contribute to the development of abnormal neurologic function.198-201 In this text, the broader term of NE is used rather than HIE or PAS. Despite the frequency with which veterinarians encounter NE, there are no direct studies in regard to the pathophysiology of NE in foals, thus the reader must acknowledge this limitation as the knowledge of this condition has been extrapolated from infants and other animal models.Pathophysiology
The exact biochemical and pathophysiologic mechanism(s) that result in NE are unknown, but at a rudimentary level, reduction in cerebral blood flow and oxygen to tissues during the antepartum (maternal hypotension, severe hypoxia, infection, placental insufficiency); peripartum (cord occlusion, premature placental separation, dystocia); or postnatal (neonatal shock, respiratory or cardiac arrest) period is believed to be a central theme in many affected patients.201 After a reversible hypoxic- ischemic brain insult, neuronal injury and/or death occurs in two phases: an acute, primary neuronal cell death followed by a delayed phase of cell death.
The primary or acute phase of injury results from reduced cerebral blood flow, which consequently decreases both oxygen and glucose delivery to the brain, thus resulting in a switch to anaerobic respiration and concomitant decrease in phosphocreatine and adenosine triphosphate (ATP) and increased lactic acid production.202,203 The decreased availability of ATP causes failure to maintain cellular integrity and homeostasis, in part due to decreased energy available to maintain both low intracellular calcium concentrations and the normal activity of cell membrane Na+∕K+ pumps.204 Not only does excessive influx of sodium into the cell cause concomitant movement of water into neurons, resulting in cell swelling and edema formation, but it also results in depolarization of neurons, which precipitates the release of the excitatory neurotransmitter, glutamate.205 Glutamate also accumulates in the extracellular space because of failure of energy-dependent glutamate uptake back into neurons.205,206 Glutamate then activates receptors within the central nervous system (CNS), resulting in sodium entry through ionotropic receptors, passive influx of chloride and water, additional influx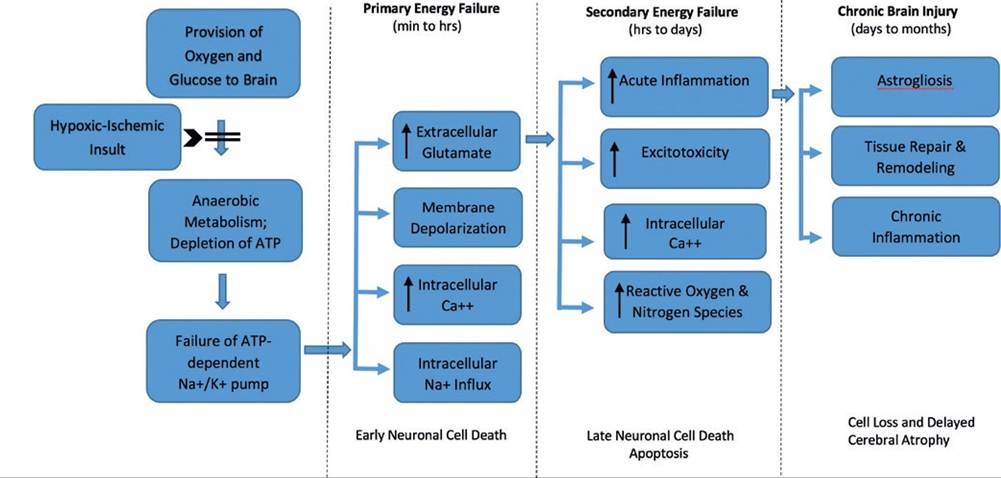
FIG. 17.7 Proposed pathophysiologic mechanisms associated with neonatal encephalopathy. Hypoxic-ischemic insult results in deprivation of oxygen and glucose to the central nervous system and cells shift to anaerobic metabolism. Reduction in adenosine triphosphate causes failure of energy-dependent cell membrane ion channels resulting in acute intracellular influx of calcium and sodium, along with cell membrane depolarization and accumulation of extracellular glutamate. This series of events can lead to cell swelling and cell death (primary energy failure). If initial insult is prolonged or severe, cascade of events can lead to excitotoxicity and increased production of inflammatory molecules and reactive oxygen species/reactive nitrogen species resulting in further cell death and apoptosis (secondary energy failure).
Chronic brain injury, characterized by astrogliosis, chronic inflammation, and tissue repair and remodeling can occur over days to months and further contribute to loss of brain cells and cerebral atrophy. (Adapted from Li B, Concepcion K Meng X, et al: Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog Neurobiol 159:50-68, 2017.)of calcium, cell swelling, and potential cell lysis.204,206 Depending on the severity of the initial insult to the brain, the acute phase of primary energy failure can result in impaired cellular integrity, disruption of the cytoskeleton and cell membrane, and ultimately cellular necrosis. Upon cell rupture, cellular contents are released, which results in additional inflammation. Microglia subsequently migrate into the area of necrosis and release further inflammatory mediators that can damage the CNS. If the insult is less severe, cells may recover or undergo apoptosis.206 Once blood flow is restored, a brief recovery or latent period occurs in which normal cerebral metabolism is reestablished.206
Conceptually, neuronal cell death as a result of a hypoxic- ischemic event and energy failure is a plausible mechanism of CNS injury and certainly contributes to the pathophysiologic mechanisms of NE (Fig. 17.7). However, studies suggest that this mechanism alone is too simplistic and continued neuronal damage occurs after the initial hypoxic-ischemic insult.205,207 The second phase of neuronal death, sometimes referred to as delayed neuronal cell death or secondary energy failure, occurs 6 to 48 hours after the initial injury and is believed to be associated with one or more of the following mechanisms: excitotoxicity, accumulation of intracellular calcium and resultant activation of numerous enzymes and pathways, reperfusion injury (oxidative stress), cytotoxic actions of activated microglia, inflammation, and apoptosis.6,199,201,203,204
Linked with hypoxia-ischemia are other damaging processes, such as the accumulation of extracellular glutamate and production of reactive oxygen species (ROS) and inflammatory mediators.
Glutamate is the predominant excitatory amino acid neurotransmitter in the brain and initiates biochemical changes through its interaction with specific receptors.6,199,206 The ionotropic receptors, so named because they are linked to ion channels, include the N-methyl-D-aspartate (NMDA) receptor, which is linked to calcium channels, and the α-amino- 3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and kainite (KA) receptors, which are linked to sodium channels.6,199,206 In the healthy individual, glutamate is released from presynaptic nerve terminals when signaled by neuronal depolarization.208 Glutamate is rapidly removed from the synaptic cleft by glutamate transporters in astroglia and converted to glutamine, which is then transported back into nerve terminals for reuse.208 Hypoxia or ischemia, as well as decreased glucose delivery to the brain, impairs normal function of astroglial glutamate transporters in the synaptic cleft, resulting in accumulation of extracellular glutamate and ensuing opening of ion channels operated by glutamate receptors.199,208 In turn, increased glutamate concentrations result in excessive calcium influx into neurons, which consequently triggers activation of lipases, proteases, and endonucleases.201 This excitotoxicity, in which excessive activation of glutamatergic neurotransmission occurs, can result in cell apoptosis and necrosis.199,208The neonatal brain is particularly vulnerable to oxidative injury owing to its high concentrations of unsaturated fatty acids, high rate of oxygen consumption, and low concentration of antioxidants.199,209 As a result of hypoxia-ischemia and/or increased intracellular calcium in neurons, a number of enzymatic pathways produce ROS through several enzyme systems including NADPH oxidase (NOX), glutathione peroxidase, cyclooxygenase, xanthine dehydrogenase, monoamine oxidase, myeloperoxidase, and nitric oxide synthase (NOS).201,209,210 Some evidence suggests that NOS and NOX, along with an oxygen- deprived mitochondrial electron transport system, comprise the major sources of ROS in the brain during a hypoxic-ischemic event.209 In addition, after a hypoxic-ischemic event, immune cells (infiltrating neutrophils, resident microglia) within the brain are stimulated and generate proinflammatory mediators and oxygen free radicals, such as superoxide anion (∙O2-), hydrogen peroxide (H2O2), hypochlorous acid (HOCl), and hydroxyl radicals (∙OH).6,209 Furthermore, NOS, specifically neuronal NOS (nNOS), which is localized with NMDA receptors and activated by calcium influx through the NMDA receptor, can produce nitric oxide (∙NO) and reactive nitrogen species (RNS); RNS can subsequently react with superoxide anion to form the powerful oxidant, peroxynitrite (ONOO-).208,211,212 Cumulatively, ROS and RNS can cause significant damage to biological proteins (membrane protein degradation), lipids (lipid oxidation), and nucleic acids (DNA degeneration).209
The inflammatory and immunologic response to a hypoxic- ischemic insult also contributes to the pathogenesis of cerebral injury.201 In the neonatal brain, an immediate innate immune response occurs within minutes of hypoxic-ischemic injury with stressed or dead neurons resulting in diffuse activation of neuroglial cells.203 Activated astrocytes, microglia, and endothelial cells then release proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1, IL-6, and IL-8.201,203 In addition, release of matrix metalloproteinases (MMPs) and proinflammatory chemokines and cytokines facilitates recruitment and activation of peripheral immune cells.203 Subsequently, activated peripheral inflammatory cells release neurotoxic agents, such as proinflammatory cytokines, NO, ROS, and myeloperoxidase, which further contribute to neuronal death, cytotoxic edema, and brain injury.203,213 Cytokines may injure white matter by inhibiting differentiation of developing oligodendrocytes, causing myelin degeneration and inducing oligodendroglial apoptosis, which further exacerbates neuronal injury.210,213 Despite the detrimental effects of the inflammatory response, however, it also serves a beneficial role by facilitating functional recovery of the CNS by eliminating cellular debris and tissue recovery.
Thus in response to hypoxic- ischemic injury, the inflammatory response attempts to remove cellular debris and promote healing of the tissues but paradoxically results in cellular injury.6Evidence of hypoxic-ischemic injury is not always identified historically or microscopically in foals with NE, and long-term neurologic deficits commonly reported in people with NE, such as cerebral palsy, visual and auditory disorders, and epilepsy, are not typically observed in foals.201 Thus alternative causes of NE have been explored. One such pathophysiologic mechanism that might play a role in a subset of foals with NE is the presence of altered concentrations of neurosteroids, which can cross the blood-brain barrier and have neuromodulatory effects resulting in clinical signs of NE in foals shortly after birth.214 In utero, the fetus maintains a quiet, suppressed, and relatively inactive state under the influence of neurosteroids, as compared with the precocious newborn foal that must be active and ambulatory shortly after birth.215 Some foals with NE have persistently increased concentrations of neurosteroids, such as plasma progestagens,200,216 and in one study, several plasma progestagens (progesterone, epitestosterone, androstenedione) were significantly increased in foals with NE when compared with foals with other disorders.214 Experimentally, intravenous infusion of allopregnenolone in a healthy newborn foal resulted in sedation and decreased responsiveness, with higher dosages of allopregnenolone resulting in dramatic neurobehavioral effects including recumbency, stupor, and unresponsiveness to the mare, environment, sound, and tactile stimulation.214 These clinical signs persisted during the CRI of allopregnenolone but waned over 15 minutes after cessation of the infusion followed by normal behavior 30 minutes post infusion.214 Allopregnenolone is believed to mediate its effects in the CNS via the GABAa receptor, and although this study only involved one foal, consideration must be granted to the possibility that persistence of fetal progestagens by the newborn foal can affect the CNS and cause signs compatible with NE in a subset of foals.77,217 Moreover, the rapid recovery from clinical signs of NE in some foals, especially in light of no obvious residual neurologic deficits, might be associated with a decline of pregnene-mediated sedative effects.214
Expanding on the theory that neurosteroids may impact the level of consciousness in newborn foals, one theory suggests that signaling of the transition from an in utero unconscious state to extrauterine consciousness may involve labor-induced physical compression (squeezing) and this compression might be associated with changes in neurosteroid concentrations.218,219 Studies investigated the use of a rope restraint device (Fig.
17.8) in neonatal foals in efforts to demonstrate that application of physical pressure via rope restraints activates the HPA axis, affects neurosteroid concentrations, and causes quiescence that is essential to the transition from fetus to newborn by preventing malposition during parturition.218,219 In one study involving eight healthy neonatal foals, a soft linen rope was secured around each foal's neck and under the shoulder and then looped around the thorax and abdomen using half-hitch knots positioned directly on the dorsal thoracolumbar area.219 The handler gently pulled on the rope from behind the foal, which produced a generalized squeezing of the foal while a second individual assisted the foal to lay down; tension on the rope was maintained for 15 to 20 minutes.218,219 During the restraint, foals assumed lateral recumbency with relaxed, somnolent behavior along with a significant decrease in heart and respiratory rate and rectal temperature. Furthermore, electroencephalography (EEG) recordings revealed patterns consistent with slow wave sleep and plasma ACTH, β-endorphin, dehydroepiandrosterone sulfate, and androstenedione concentrations significantly increased during restraint, compared with pre-restraint values.219 A follow-up survey involved equine clinicians that collected observational data from foals with NE that were treated with standard medical therapy with or without the inclusion of the rope restraint/squeeze method.218 The survey inquired about medical treatments and a response to medical treatment only (nonsqueezed group) versus the squeeze procedure with or without medical therapy (squeezed group). There were 51 respondents to the survey providing information on 195 foals with NE (108 nonsqueezed; 87 squeezed). In this survey, foals that received the squeeze procedure with or without medical therapy had significantly faster recovery rates, were 3.7 times more likely to recover faster, and were 15 times more likely to recover in less than 1 hour than foals that were not squeezed. The authors concluded that foals that were squeezed for 20 minutes had significantly faster and higher recovery rates than foals that did not receive the procedure. There were several limitations to this study, but the authors proposed that the pressure from the birth canal that the fetus is exposed to during stage 2 of labor might influence the balance of neurosteroids and play a role in the neonatal transition from neuroinhibition 218220221
FIG. 17.8 Foal fitted with a soft linen rope restraint device. Squeeze effect generated by a person who stands behind the foal and gently pulls on the rope; an assistant helps the foal to lie down. Completion of the rope squeeze after 15 to 20 minutes.
to neuroactivatton.218,22°,221 Thus simulation of stage 2 labor with the rope restraint may reset the balance of these neurosteroids and expedite recovery from NE and facilitate normal behavior in the neonatal foal.
Yet another alternate theory by which foals may develop NE outside the framework of a hypoxic-ischemic insult involves the inflammatory response in the neonatal brain associated with chorioamnionitis (placentitis), maternal and fetal inflammation, and resultant injury to the fetal/neonatal brain.,, In health, the chorioallantois has properties of an immunologically privileged site in order to protect the allogenic fetus from rejection by the mother. When an infectious agent invades the placental membranes, both a maternal and fetal inflammatory response, characterized by release of proinflammatory and inhibitory cytokines and chemokines, can occur. The initial response to intrauterine infection involves leukocytes derived from the maternal circulation (maternal inflammatory response). Subsequently, intrauterine inflammation also involves leukocytes derived from the fetal vessels and systemic activation of the fetal innate immune system (fetal inflammatory response syndrome, FIRS).223 Both maternal and fetal inflammatory responses are typified by production of inflammatory mediators (IL-1, IL-6, TNF-α) and activation of neutrophils and other components of the innate immune system.222,223 Although not fully understood, the FIRS may induce cerebral white matter injury, intraventricular hemorrhage, and/or periventricular leukomalacia, manifesting clinically as neurologic deficits. Proposed mechanisms by which this occurs includes cerebral hypoperfusion and ischemia, activation of the coagulation cascade resulting in capillary thrombosis, and necrosis of white matter and/or increased permeability of the blood-brain barrier, which potentially allows direct passage of microbes and cytokines into the CNS.222,223 Other body systems that may be impacted by FIRS include the hematopoietic system, thymus, adrenal glands, skin, kidneys, heart, and lung. Equine studies directly linking placentitis, FIRS, and brain injury are lacking, but placentitis is a noted risk factor for the development of NE in foals and a study investigating placentitis in mares demonstrated significantly more expression of mRNA for IL-6 and IL-8 from the cervical star region in mares with experimentally induced Streptococcus equi ssp. Zooepidemicus placentitis when compared with healthy pregnant mares.6,224 Thus it is plausible, but unsubstantiated, that placentitis and FIRS may play some part in the pathophysiologic development of NE in foals.
Risk Factors and Clinical Signs
Recognized risk factors for NE in infants include maternal systemic hypotension, maternal cardiac arrest, clotting of placental arteries, placental abruption, uterine rupture, and inflammation.203 In comparison, risk factors in horses associated with NE include dystocia, induced parturition, Cesarean section, placentitis, premature placental separation, severe illness, post-term pregnancy (fescue toxicity), and maternal hypotension (endotoxemia, hemorrhage, anemia, severe respiratory disease).225-228 Other factors, such as meconium aspiration, twin foals, fetal infection, congenital malformations, and umbilical cord accidents, may also be risk factors for the development of NE.225-228 In a retrospective study involving 79 neonatal foals with a primary diagnosis of NE, the presence of placental abnormalities (55% of foals), gestational problems (21%), premature placental separation (34%), and dystocia (30%) were reported.197 Conversely, foals with NE may have no reported problems during gestation or parturition.
In regard to clinical signs, affected foals can be born demonstrating abnormal behavior or, alternatively, demonstrating clinical signs of NE 12 to 48 hours after birth. This temporal observation of delayed clinical signs may reflect the biphasic acute and delayed injury to the CNS discussed in the proposed pathophysiology. Clinical signs are quite variable and range in severity to include alterations of consciousness (stupor, somnolence, difficult to arouse, coma); weakness (hypotonia); inability to suckle; lack of affinity for the dam; inability to find the udder; inappropriate nursing behavior; localized or generalized seizures; tongue protrusion or weak tongue tone; star gazing; loss of recognition of the environment; abnormal vocalization; dysphagia; central blindness; irregular respiratory pattern (apnea, abnormally slow respiratory rate); and pro- 625229230
prioceptive deficits. ,25,, In a retrospective study involving 94 foals, the most frequent reported clinical signs included abnormal udder seeking (59%), abnormal suckle (55%), inability to stand (42%), abnormal gastrointestinal motility (37%), abnormal consciousness (34%), and seizure activity (22%).230 In another retrospective report, dysphagia was observed in 16 neonatal foals, and of these 16 foals, 6 foals (38%) had a clinical diagnosis of NE; 15/16 foals were discharged from the hospital (median 6.3 days; range 0 to 22 days), with 14/15 nursing from the mare with no evidence of dysphagia. Clinical signs reflective of other body systems that can be impacted by hypoxic-ischemic injury, such as the renal, gastrointestinal, cardiac, and pulmonary system, also can be present. Moreover, clinical signs of other concurrent diseases that occur with NE, such as sepsis, pneumonia, prematurity/dysmaturity, patent urachus/omphalitis, limb deformity, colic, and uroperitoneum, may also be observed.230
Diagnosis
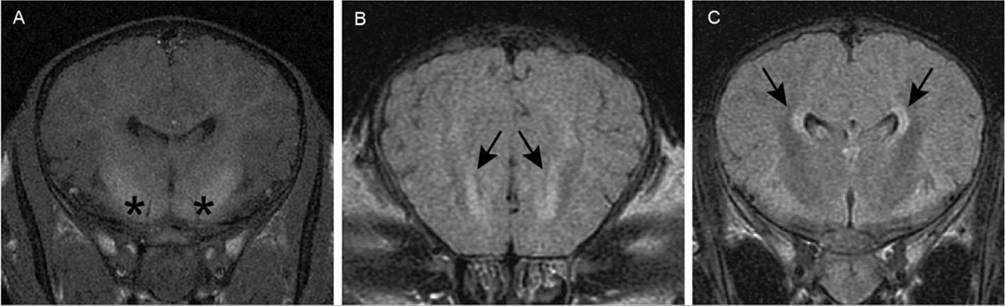
FIG. 17.9 Magnetic resonance imaging scans of the brain of a 5-day-old Thoroughbred filly with symmetric abnormalities in the cerebrum. A, T-1 transverse image through the cerebral hemispheres immediately rostral to the pituitary. Asterisks indicate regions of hyperintensity of basal nuclei. B, T2-weighted (T2W) fluid-attenuated inversion recovery (FLAIR) transverse image through rostral limit of lateral ventricles. Arrows indicate areas of hyperintensity associated with rostral poles of the ventricles. C, T2W FLAIR image through cerebral hemispheres immediately rostral to pituitary (same level as image A). Arrows indicate regions of hyperintensity adjacent to lateral ventricles. (From Wong DM, Jeffery N, Hepworth-Warren KL: Magnetic resonance imaging of presumptive neonatal encephalopathy in a foal. Equine Vet Educ 29:534-538, 2017).
and disassociation from the mare, an MRI of the brain was performed because of worsening clinical signs of NE (i.e., seizures).239 MRI revealed symmetric, poorly delineated areas of high signal intensity (compared with gray matter regions elsewhere) involving the basal nuclei (accumbens, putamen, claustrum); ventral thalamic nuclei; and rostral aspect of the ventral midbrain (in the region of the substantia nigra) on T1-weighted (T1-W) images (Fig. 17.9).239 Hyperintensity associated with the margins of the lateral ventricles were noted on T2-weighted (T2-W) and T2-W fluid-attenuated inversion recovery (FLAIR) images.239 The lesions noted in this foal were similar to those described in infants with HIE.234,235 Whether or not MRI and its associated risks of general anesthesia and added client expense is a feasible diagnostic test in foals with NE remains to be determined, but imaging studies might help elucidate the pathophysiology of NE in foals and guide prognostication.
The use of cranial US has also been used in infants to help identify neonates with HIE, as well as other disorders, such as cranial hemorrhage, congenital structural abnormalities of the cerebrum, and other causes of fetal brain injury.232 Abnormalities associated with HIE in infants that were identified with cranial US included changes in ventricular size, periventricular echogenicity, increased echogenicity in the thalami, extra-axial fluid collection, and intracranial hemorrhage.232 Although cranial US was able to identify all infants with a poor outcome, MRI was better at detecting changes associated with HIE.240 One study in foals has examined and described ultrasonographic measurements of a variety of variables viewed from the atlanto-occipital (AO) space in healthy neonatal foals and foals with NE.241 In that study, an 8-MHz microconvex transducer was used to image the AO space using a depth of 6 to 8 cm. Specifically, the transducer was placed immediately cranial to the wings of the atlas to obtain a transverse image and then rotated 90 degrees to obtain a longitudinal image.241 Measurements of the height of the spinal cord, depth of dorsal subarachnoid space (mid-AO space), maximal depth of the dorsal subarachnoid space (cranial-AO space), and dorsoventral diameter of the ventral spinal artery were collected from longitudinal images (Fig. 17.10).241 On transverse images, measurements of the height, width, and cross-sectional area of the spinal cord and spinal canal were taken along with the depth of the dorsal subarachnoid space. In addition, ratios of the spinal canal to spinal cord height measurements, spinal canal to cord width measurements, and spinal canal to cord cross-sectional area measurements were calculated from transverse images. When comparing healthy foals with foals with NE, the dorsoventral diameter of the ventral spinal artery (longitudinal images) and absolute measurements of the spinal cord height, width, and cross-sectional area (transverse images) were all significantly smaller in foals with NE.241 Moreover, ratios of spinal canal to spinal cord width and cross-sectional area were significantly larger in foals with NE when compared with healthy foals. One possible explanation for the reduced spinal cord dimensions in foals with NE is related to elevated intracranial pressure; however, the clinical application, utility, and reliability of ultrasonographic examination of the AO region in foals suspected to have NE is unknown and requires further study.241
EEG is the graphic recording of the rhythmic bioelectrical activity that arises predominantly from the cerebral cortex and has been used in people and, to a lesser extent, horses as a screening procedure in patients with intracranial neurologic disorders.242 There is a lack of a single accepted classification system in regard to EEG interpretation in infants evaluated for NE, so a good degree of variation in terms of EEG definitions and grading schemes are published within the litera- ture.243,244 Commonly reported abnormal EEG findings in infants with NE include frequent spikes, positive temporal sharp waves, polyphasic sharp waves, mild voltage depression, persistent discontinuity, burst suppression, and modified burst suppression pattern.243,244 In one study involving a 1-day-old filly, later confirmed to have histopathologic abnormalities compatible with NE, EEG recordings revealed diffuse high- voltage slow waves and focal discrete paroxysmal discharge activity, as well as multifocal paroxysmal activity. The practical application of EEG in equine medicine remains limited because of the restricted availability of equipment and trained personnel to perform and interpret the EEG.242
Finally, a variety of blood parameters also have been examined in foals with NE. In one study, 32% of foals with NE had increased serum creatinine concentration and 61% of affected foals had increased activity of serum creatine kinase.197 Another retrospective study investigating spurious hypercreatinemia noted that of 28 foals with spurious hyper- creatinemia (mean creatinine concentration 13.6 mg/dL), 20 foals (71%) had a clinical diagnosis of NE.245 However, elevations in serum creatinine and creatine kinase can be associated with a number of other disease processes in neonatal foals. A more recent equine study measured plasma concentrations of ubiquitin C-terminal hydrolase 1, a biomarker for brain injury, and noted significantly higher values in foals with NE (mean 6.57 ng/mL; range 2.35 to 11.90 ng/mL) when compared with
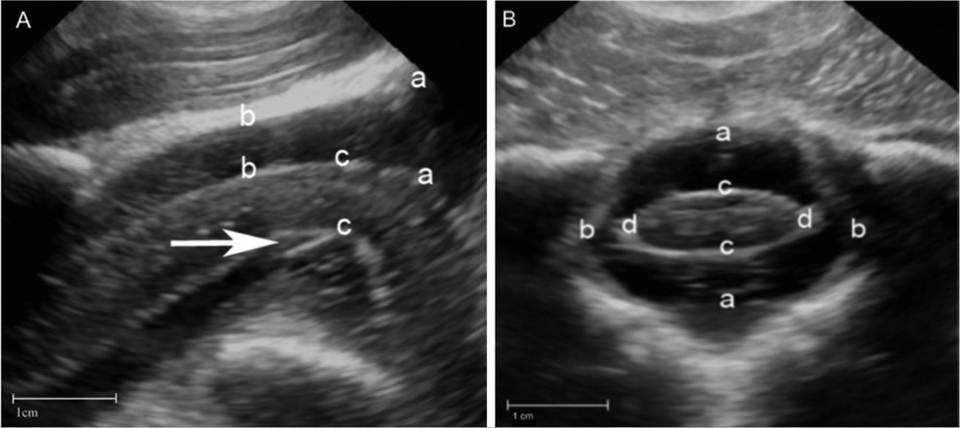
FIG. 17.10 Ultrasonographic images of the atlanto-occipital region in a 1-day-old Thoroughbred foal with neonatal encephalopathy. A, Longitudinal ultrasound image (cranial to right of image). Measurements of the maximal depth of the dorsal subarachnoid space (cranial atlanto-occipital space) (a to a), dorsal subarachnoid space (mid atlanto-occipital space) (b to b), height of the spinal cord (c to c), and dorsoventral diameter of the ventral spinal artery (arrow). B, Transverse ultrasound image (right side of foal to right of image). Measurements of the spinal canal height (a to a) and width (b to b), spinal cord height (c to c), and width (d to d), and dorsal subarachnoid space (dorsal a to dorsal c). (From Mackenzie CJ, Haggett EF, Pinchbeck GL, et al. Ultrasonographic assessment of the atlanto-occipital space in healthy Thoroughbred foals and Thoroughbred foals with neonatal maladjustment syndrome. Vet J 223:55-59, 2017.).
healthy foals (median 2.53 ng/mL; range 1.4 to 4.01 ng/mL). Currently, measurement of this biomarker is not readily available for clinical use.246
At present, practicality dictates that NE in foals be based primarily on clinical impression, founded on historical information, neurologic examination, compatible clinical signs, and exclusion of other disease processes. In foals, NE may occur as a primary problem or may be coupled with failure of passive transfer of maternal antibodies, sepsis, musculoskeletal abnormalities, prematurity/dysmaturity, and other congenital disorders. Thus the clinician should perform diagnostics, such as a complete blood count, serum biochemistry profile, arterial blood gas analysis, blood culture, urinalysis, and assessment of passive transfer of maternal antibodies, to establish a complete assessment of the foal.6
Treatment
In equine medicine, the core component of treatment of NE is appropriate supportive and nursing care. This entails maintenance of adequate blood pressure and hydration, meeting caloric needs, facilitation of gas exchange, prevention of skin sloughing and ulceration by keeping the foal clean and dry, and prevention or treatment of infection. There are varied opinions on appropriate intravenous maintenance fluid rates for neonatal foals, but one approach is the Holliday-Segar method2 (see previously and Chapter 44); the clinician must also consider renal function in calculating IV fluid rates as the kidneys can sustain injury from hypoxic-ischemic insult. Maintenance of adequate blood pressure and perfusion is also important for supporting cerebral (and other organ) perfusion and avoiding further ischemic injury., Identifying any underlying cause(s) of hypotension, coupled with cautious use of IV fluid therapy and vasopressors or inotropes, might be necessary. Likewise, slow and judicious introduction of enteral milk feedings should be implemented as the gastrointestinal tract can be impaired from hypoxia-ischemia. Foals should be evaluated for gastric reflux before feeding. A conservative rate of 5% of the foal's body weight of mare's milk divided over a 24-hour period (i.e., 50 kg ? 0.05 = 2.5 L mare's milk delivered every 2 hours over a 24-hour period = 100 mL every 2 hours) is a reasonable starting point, while monitoring for any clinical signs of indigestion (gastric reflux, colic). Milk supplementation should be increased gradually to a total of 20% to 25% of the foal's body weight per day over the next several days if the foal's gastrointestinal tract is functioning normally. Alternatively, or in addition to milk feedings, IV glucose supplementation can be used to provide another source of calories. A rate of 5 mg/kg/min of glucose has been suggested as an initial IV glucose CRI flow rate based on placental blood flow in late gestation (6.8 mg/kg/min).9 This equates to an hourly flow rate of 3 mL/kg (150 mL/hr for a 50-kg foal) of a 10% glucose solution. Blood glucose concentration must be monitored and fluid rate adjusted routinely to avoid hyperglycemia or hypoglycemia. Moreover, as energy depletion plays a role in the development of NE, coupled with the fact that neonates have minimal energy reserves, it is important to maintain the blood glucose concentration within the reference range.247,248 If enteral feedings cannot be initiated in the first 24 to 48 hours, consideration of partial (PPN) or total parental nutrition (TPN) should be given. An arterial blood gas sample should be performed to gauge pulmonary function and supplemental intranasal oxygen provided if necessary. Furthermore, foals should be maintained in sternal recumbency where possible to facilitate adequate ventilation of both left and right lung lobes. If hypoventilation and hypercapnia are observed, doxapram (0.02 to 0.05 mg/kg/hr) can be administered as a CRI as a respiratory stimulant.249 Although doxapram was found to be more effective at lowering PaCO2 in foals with NE, caffeine can be administered per os (loading dose 7.5 to 12 mg/kg, followed by daily dose 2.5 to 5 mg/kg) if a CRI is not feasible. Provision of soft bedding with clean dry blankets will help prevent skin trauma and ulceration. Assessment of passive transfer of maternal antibodies should be evaluated as many NE foals fail to ingest enough colostrum and IV administration of equine plasma can be necessary. The rope restraint squeeze method can also be used in efforts to alter neurosteroid concentrations, and administration of broad-spectrum antimicrobials is indicated to treat or prevent sepsis.219 Evaluation and treatment of other body systems, particularly the gastrointestinal and renal system, which are predisposed to hypoxic-ischemic insult, should be considered.
Many foals with NE will respond to the aforementioned treatment suggestions within 3 to 5 days and have a complete recovery. However, a few specific therapies should also be considered with more complicated cases of NE. Moreover, numerous medications have been suggested, mainly used in infants with NE, that target specific pathophysiologic mechanisms that may be involved with the development of NE. Importantly, seizures should be treated immediately if observed, as ongoing seizure activity increases cerebral oxygen consumption and can further contribute to neuronal damage. Intravenous diazepam (0.1 to 0.2 mg/kg) typically controls seizures in the neonatal foal and can be given intermittently if the incidence of seizures is infrequent. An alternative benzodiazepine that can be used for seizures is midazolam given as a single dose (0.04 to 0.1 mg/kg IV) or a CRI (0.02 to 0.06 mg/kg/h IV) to control intractable seizures.250 If seizure activity persists, phenobarbital (2 to 10 mg/kg IV over 15 minutes, then 5 mg/kg PO q12h) may be necessary. Side effects of phenobarbital, such as mild sedation, ataxia, and hypothermia, can be observed. Intravenous anesthesia with propofol can be required for several hours to manage intractable cases of status epilepticus.25 Pharmacokinetics of levetiracetam, a novel antiepileptic drug used in people and dogs, has recently been investigated in healthy neonatal foals, but exact therapeutic ranges for this drug have not been established in horses.251 On the basis of this study, levetiracetam administered at a dose of 32 mg/kg by mouth twice daily was predicted to maintain plasma concentrations above target concentrations for other species. Published use of levetiracetam in foals with NE is not available but might have potential for seizure control in future cases, although adjustment of dose and frequency of administration may be required in the clinical setting.251 Hyperbaric oxygen therapy has been suggested as a potential therapy for NE with proposed benefits including reduced apoptosis, promotion of neuronal stem cell proliferation, enhancement of oxygen radical scavengers, increased oxygen delivery to the brain, and increased activity of superoxide dismutase.6,210,252 Hyperbaric oxygen therapy has been used in a limited number of infants and foals with NE and has been purported to have beneficial effects.253-255
Medications that combat edema and oxidative damage have been used in foals with NE but have not undergone clinical trials or received scientific scrutiny; thus their effectiveness has been debated. Nonetheless, some clinicians have used mannitol (0.5 to 2.0 g/kg as a 20% solution over 20 minutes, IV q12-24h) or furosemide (1 mg/kg IV q12-24h) in efforts to reduce cerebral edema. Dimethyl sulfoxide (0.5 to 1 g/kg IV as a 10% solution given over 30 minutes, q12-24h) has also been used to treat cerebral edema and for its purported free radical scavenging abilities. Other putative therapeutics acting as free radical scavengers or antioxidants administered to foals with NE include thiamine (5 to 10 mg/kg IV or SC q24h), vitamin E (20/kg IU/kg, SC, or PO q24h), and vitamin C (100 mg/kg/day, IV). Allopurinol is a medication that inhibits xanthine oxidase, the enzyme responsible for production of superoxide radicals during reperfusion injury , ; a dosage of 6 mg/kg PO once was used in a foal in conjunction with other treatments.209a
Accumulation of glutamate and excitotoxicity is another component of the pathophysiology of NE, and medications that help reduce the impact of excess glutamate have been investigated. Magnesium sulfate (MgSO4) prevents calcium entry into the cell by noncompetitive voltage-dependent inhibition of NMDA receptors.201 This in turn blocks the release of glutamate and antagonizes the influx of calcium.201,209 In addition, MgSO4 reduces secondary inflammation-induced injury via cell membrane stabilization and inhibition of free radical formation.201 Rodent models have demonstrated that MgSO4 can be strongly protective against hypoxic-ischemic injury if given before or shortly after a hypoxic-ischemic insult.208 A dose of 0.05 g/kg/h of MgSO4 for the first hour, followed by 0.025 g/kg/h IV as a CRI has been suggested as a treatment for NE in foals.256
Therapeutic hypothermia is currently part of the standard treatment for neonatal infants with moderate to severe NE aiming for cooling to commence within 6 hours of hypoxic- ischemic insult to a temperature of 32°C to 34°C for 48 to 72 hours, followed by a gradual rewarming over 12 hours.201 A number of studies have found improved neurodevelop- mental scores in NE infants treated with hypothermia with proposed protective mechanisms of hypothermia including reduced cerebral metabolism, suppression of caspase-3 enzyme and microglial activation, prevention of activation of NMDA receptors, reduction of proinflammatory cytokines, and preservation of lipoprotein membranes.201,203,229,257,258 Published reports of therapeutic hypothermia in neonatal foals with NE are not available. Other medications reported to be used in infants with NE or animal models, alone or in conjunction with hypothermia therapy, include melatonin (antiinflammatory, antiapoptotic, antioxidant properties); erythropoietin (antiinflammatory, antiapoptotic factors, angiogenesis, neurogenesis properties); N-acetylcysteine (clears free radicals, restores glutathione levels, antiinflammatory, reduces NO production); topiramate (glutamate receptor [AMPA and kainite] inhibition, blocks Na+ channels and activates Ca+ currents); desferrioxamine (reduction of free radical production by chelating iron); xenon (NMDA receptor antagonist, antiapoptotic factors); edaravone (free radical scavenger); and stem cells and IL-1 receptor antagonists.201,203,208,209,229 These therapies and medications require further research in infants and have not been currently used for the treatment of NE in neonatal foals.
Prognosis and Pathologic Findings
In general, foals with a primary diagnosis of NE without complicating factors have a good prognosis for both survival and athletic function with reported survival rates ranging from 70% to 80%.150,197,225-228,230 The prognosis is poorer in foals that demonstrate clinical signs at the time of birth, have evidence of brainstem or spinal cord involvement, have multiple organs affected by hypoxic-ischemic injury, have complicating factors (e.g., sepsis), remain comatose or are difficult to arouse, show no improvement in neurologic function in the first 5 days, or have severe recurrent seizures.6,225,227 Recumbency was significantly associated with nonsurvival in one retrospective study,230 and in a different retrospective study of 78 foals with NE, neurologic signs lasted 1 day in 29 foals (29/59 foals that survived; 49%), 2 days in 17 foals (17/59; 29%), 3 days in 8 foals (13.5%), 4 days in 4 foals (6.8%), and 5 days in 1 foal (1/59; 1.7%); 19/78 foals (24%) did not survive.197 Rare longterm neurologic deficits include inability to suckle from the mare, prolonged visual impairment, residual spasticity, recurrent seizures, and docility as adults.197,227
Four major categories of neuropathology are noted in infants with NE: selective neuronal necrosis, parasagittal cerebral injury, periventricular leukomalacia, and focal ischemic brain necrosis.229 Histopathologic findings reported in foals with NE are likely skewed toward more severe cases of NE as the majority of foals survive and accordingly do not undergo microscopic evaluation of the CNS. In one retrospective study involving 11 available microscopic descriptions of foals with NE, 10 of 11 foals had histologic evidence of neuronal necrosis and degeneration within the CNS consistent with ischemia.230 In another study involving 18 foals with NE, necrosis of the cerebral cortex and local hemorrhage was noted in 9/18 foals,259 whereas neuronal necrosis of the gray matter of the cerebral cortex, caudate nuclei, thalamus, hippocampus, cerebellar cortex, and medulla oblongata was reported in 3 foals with NE in another report.246