One of the Most Important Clinical Uses of Glucocorticoids Is the Suppression of the Inflammatory Response
Glucocorticoids have valuable clinical effects, particularly the inhibition of the inflammatory response, including the prevention of capillary dilation, extravasation of fluid into tissue spaces, leukocyte migration, fibrin deposition, and connective tissue synthesis.
Whereas the process of inflammation is important for the walling off and destruction of systemic noxious agents, the end response is often the replacement of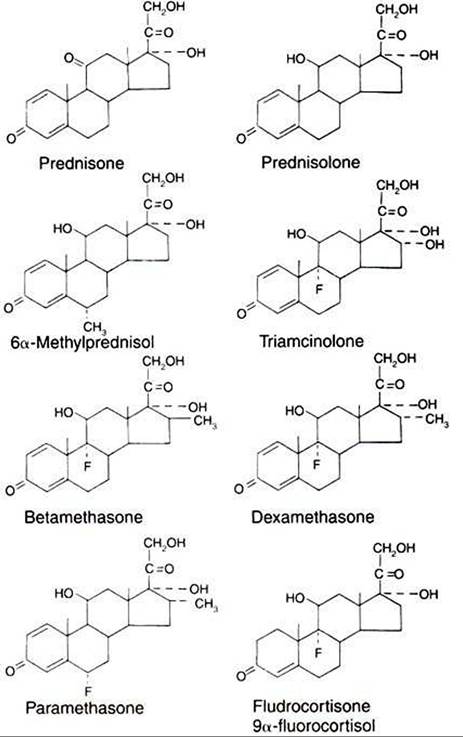
FIGURE 34-11 Chemical structures of some clinically useful glucocorticoid analogues. (From Martin CR: Endocrine physiology, London, 1985, Oxford University Press.)
functional tissue with fibrous connective tissue, with a resultant loss of function. For example, inflammatory processes in the mammary gland often result in the isolation of the injurious agent by the laying down of connective tissue as a part of the defense mechanism; however, the gland may lose much of its functional capacity as a result. Administration of glucocorticoids, in conjunction with antibiotic therapy, can help reduce the loss of functional tissue by inhibiting the development of connective tissue. Figure 34-11 shows the chemical structures of some of the synthetic glucocorticoids used in clinical practice.
One of the ways in which glucocorticoids inhibit the inflammatory response is through the inhibition of the formation of substances that promote inflammation. Glucocorticoids inhibit the synthesis of inflammatory mediators, such as prostaglandins, thromboxanes, and leukotrienes, that arise as a result of arachidonic acid metabolism. This effect is mediated through the stabilization of lysosomal membranes and the prevention of phospholipase A2 activation. Glucocorticoids are also used to inhibit allergic reactions. This action occurs through the inhibition of the release of certain biogenic amines, such as histamine, from the granules of mast cells.
Hyperadrenocorticism
Hyperadrenocorticism (Cushings syndrome) in the dog may be caused by a pituitary tumor, pituitary hyperplasia, adrenal tumors, adrenal hyperplasia, or nonendocrine tumors (usually of the lung), or it may be iatrogenic. Approximately 85% of dogs with Iiyperadrenocorticism have pituitary gland-dependent disease, whereas 15% exhibit adrenal tumors. Hyperadrenocorti- cism is a disease of middle-aged and older dogs (7-12 years). Breeds typically affected by pituitary-dependent hyperadreno- corticism include miniature poodles, dachshunds, boxers, Boston terriers, and beagles. Adrenal tumors are seen more frequently in large-breed dogs, and there is a predilection for females (3:1 ratio with males). Hyperadrenocorticism is a rare endocrine disorder of cats and is usually pituitary in origin in that species.
The most common clinical signs associated with canine hyperadrenocorticism are polydipsia, polyuria, polyphagia, heat intolerance, lethargy, abdominal enlargement or “potbelly,” panting, obesity, muscle weakness, and recurrent urinary tract infections (UTIs). Dermatological manifestations of canine hyperadrenocorticism can include alopecia (especially truncal), thin skin, phlebectasias, comedones, bruising, cutaneous hyperpigmentation, calcinosis cutis, pyoderma, dermal atrophy (especially around scars), seborrhea, and secondary demodicosis. Thin skin is the hallmark of feline hyperadrenocorticism. Cats with Cushing’s syndrome develop such severe thinning of the epidermis that they may incur open wounds just by grooming themselves.
Attempts to diagnose hyperadrenocorticism can be challenging. Uncommon clinical manifestations of hyperadrenocorticism in dogs can include signs such as hypertension, congestive heart failure, bronchial calcification, pulmonary thromboembolism, polyneuropathy, polymyopathy, pseudomyotonia, behavioral changes, and blindness. Evidence of increased collagenase activity caused by hypercortisolemia may result in nonhealing corneal ulceration and bilateral cranial cruciate rupture (in small dogs).
Unusual reproductive signs may include testicular atrophy, prostatomegaly in castrated male dogs, clitoral hypertrophy, and perianal adenoma in females or castrated males.Serum chemistry abnormalities associated with hyper- Cortisolemia in dogs include increased serum activities of alkaline phosphatase and alanine aminotransferase, hypercholesterolemia, hyperglycemia, and decreased BUN. The hemogram is often characterized by evidence of erythroid regeneration (nucleated red blood cells) and a classic “stress leukogram.” Basophilia is occasionally observed. Many dogs with hyperadrenocorticism have evidence of UTI without pyuria. Proteinuria resulting from glomerulosclerosis is also common. Urine specific gravity is usually decreased and may be hyposthenuric. Thyroid status is often affected in animals with hyperadrenocorticism, as evidenced by (1) decreases in TT4 and TT3 caused by euthyroid sick syndrome and (2) a response to TSH stimulation that is attenuated as a result of overcrowding of pituitary thyrotrophs by adrenocortico- trophs. Overt diabetes mellitus may result from the insulin antagonism caused by hypercortisolemia in about 15% of dogs with hyperadrenocorticism and 85% of cats with hyperadrenocorticism. Conversely, hyperadrenocorticism can be a cause of insulin resistance and poor glycemic control in diabetic animals.
'Γhe diagnosis of hyperadrenocorticism should be based on suggestive clinical signs and supporting minimal database abnormalities (e.g., high serum cholesterol, increased serum alkaline phosphatase activity) and confirmed by an appropriate screening test. If screening test results are inconclusive, the dog should be retested at a later date (3-6 months) rather than be subjected to treatment without a definitive diagnosis.
Screening tests for hyperadrenocorticism, such as the low- dose dexamethasone suppression (LDDS) test and the corticotropin stimulation test, work on the principle of suppression or stimulation of the pituitary-adrenal axis.
In the case of the LDDS test, dexamethasone is administered at a low dosage to cause negative feedback to the pituitary gland. In a normal animal, this negative feedback results in a decrease in endogenous corticotropin secretion and a resultant decrease in circulating cortisol concentrations. Dexamethasone is the only synthetic corticosteroid that does not cross-react with the cortisol assay. The corticotropin stimulation is used to determine the extent of adrenal enlargement. Adrenal glands that are enlarged because of chronic pituitary stimulation by corticotropin or that are neoplastic show an exaggerated response to exogenous corticotropin.The LDDS test has traditionally been the screening test of choice for canine hyperadrenocorticism. It is sensitive (92%- 95%); only 5% to 8% of dogs with pituitary dependent hyperadrenocorticism exhibit suppressed cortisol concentrations at 8 hours (i.e., 5%-10% false-negative results). In addition, 30% of dogs with pituitary-dependent hyperadrenocorticism exhibit suppression at 3 or 4 hours, followed by “escape” of suppression at 8 hours; this pattern is diagnostic for pituitarydependent disease and makes further testing unnecessary. The major disadvantage of the LDDS test is the lack of specificity in dogs with nonadrenal illness. It is recommended that a dog be allowed to recover from the nonadrenal illness before being assessed for hyperadrenocorticism with the LDDS test.
Mineralocorticoids
The mineralocorticoids, produced in the outer zone (zona glomerulosa) of the adrenal cortex, have surprisingly different functions compared with glucocorticoids; the functions are surprising because both types of hormones are produced by tissues that are part of the same gland. As indicated previously, electrolyte balance and blood pressure homeostasis represent the principal physiological effects of mineralocorticoids (Table 34-4). These actions are carried out at the level of the distal tubules in the kidney. The effect of the mineralocorticoids is to promote retention of sodium and secretion of potassium and hydrogen.
Γhe cellular response to mineralocorticoids is to synthesize a protein that increases the per-Table 34-4
Mineralocorticoid Effects and Target Tissues
| Effect | Site of action |
| Stimulates Na+ reabsorption | Kidney, salivary glands, sweat glands |
| Stimulates K+ excretion | Kidney, salivary glands, sweat glands |
| Stimulates H+ excretion | Kidney |
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
meability of the luminal cell surface to sodium influx from the renal filtrate and increases sodium/potassium-adenosinetri- phosphatase (Na+,K+-ATPase) activity in the contraluminal cell surface, which allows movement of Na+ out of the cell into the interstitial tissue (Figure 34-12).
The control of secretion of K+ by mineralocorticoids is passive in the sense that K+ is retained in the renal filtrate to maintain the osmolality of urine. However, evidence suggests that mineralocorticoids have an effect on Na+ secretion that is
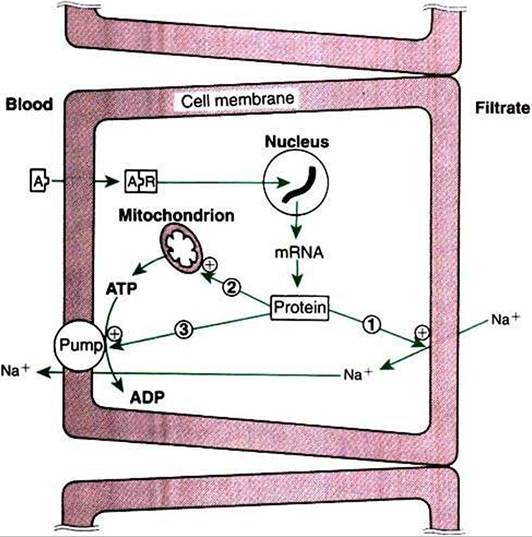
FIGURE 34-12 Mechanisms of action of aldosterone on sodium transport in the renal tubular cell.The numbered arrows indicate the three putative sites of action of aldosterone: 7z increasing the permeability of the luminal membrane to sodium; 2, increasing mitochondrial adenosine triphosphate (ATP) production; and (3) increasing Na+,K+-ATPase activity in the contraluminal membrane. Plus signs indicate stimulation. At Aldosterone; ADPt adenosine diphosphate; mRNAt messenger ribonucleic acid; Rt receptor.
(From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)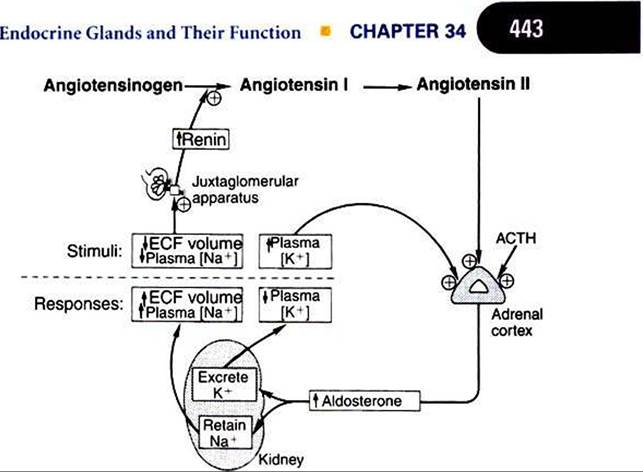
FIGURE 34-13 Regulation of aldosterone secretion by the zona glomerulosa of the adrenal cortex. Plus signs indicate stimulation. ACTHt Corticotropin (adrenocorticotropic hormone); ECFt extracellular fluid. (From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
independent of Na‘ retention. The secretion of K' continues to be influenced by mineralocorticoids after mineralocorticoid administration, whereas Na+ retention decreases within a few days.
In situations of excessive mineralocorticoid production, the effects of increased Na' retention are to increase the extracellular fluid volume and to cause hypertension; conversely, low blood pressure (hypotension) occurs as a result of inadequate secretion of mineralocorticoids. Hypersecretion of mineralocorticoids can also lead to excessive hydrogen ion (H+) loss and metabolic alkalosis, whereas hyposecretion can result in increased retention of H* and metabolic acidosis.
The regulation of mineralocorticoid secretion, in contrast to glucocorticoid secretion, is not controlled by tropic hormones from the pituitary gland (Figure 34-13). In the case of mineralocorticoids, the main controlling factors are produced in the target organ, the kidney. Cells in the juxtaglomerular apparatus of the kidney produce an enzyme, renin, in response to decreases in blood pressure. Renin acts on αngiotensinogent an α2 globulin produced by the liver and present in the circulation, and this results in the production of angiotensin It a decapeptide. Angiotensin I is further hydrolyzed to angiotensin IIt an octapeptide, by angiotensin-converting enzyme. Angiotensin Il stimulates the zona glomerulosa to produce mineralocorticoids. Angiotensin II also increases peripheral resistance of the blood vascular system by causing vasoconstriction of smooth muscle of the blood vessels. Angiotensin II, if present on a long-term basis, also increases the size of the zona glomerulosa.
Evidence indicates that cells of the macula densa, groups of specialized cells located at the origin of the kidney’s distal tubule (Figure 34-14), exert control on the renin-angiotensin system. This is done through the sensing of changes in Na+
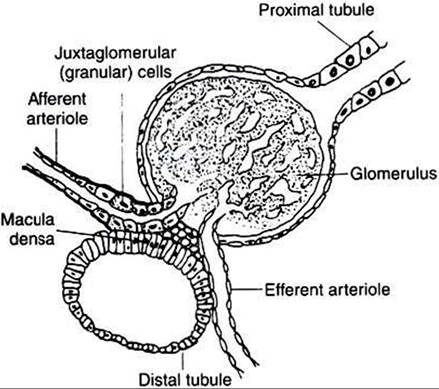
FIGURE 34-14 Diagrammatic representation of juxtaglomerular apparatus. (From Martin CR: Endocrine physiology, London, 1985, Oxford University Press.)
concentrations in tissue fluids; increased Na, results in decreased renin release» and decreased Na+ results in increased renin release. In either case, the change produced tends to restore the mineralocorticoid concentrations to normal. In addition to the effect of sodium, the macula densa may control changes in the renin-angiotensin system through the sensing of changes in chloride ion (CΓ) concentrations in tissue fluids.
Another major regulatory factor in the control of mineralocorticoid secretion is the blood potassium concentration. An increase in K+ concentration stimulates the zona glomerulosa to secrete mineralocorticoids, whereas a decline in K+ has the opposite effect. This stimulation is independent of the reninangiotensin system.
It has been thought that corticotropin has minimal effect on control of the zona glomerulosa, because experimental studies showed that hypophysectomy has little effect on the zona glomerulosa. More recent studies have shown that cells of the zona glomerulosa have receptors for corticotropin, and corticotropin may play some role, although minor, in the control of mineralocorticoid secretion.
In contrast to the sodium-conserving effect of mineralocorticoids, the 28-amino acid atrial natriuretic peptide (ANP) reduces Na+ retention by the kidneys. ANP also causes peripheral vasodilation and thus a lowering of blood pressure. ANP may inhibit the production of mineralocorticoids and renin as well. ANP is produced by cells of the cardiac atria, but it is also produced in other sites, including the brain.
Hypoadrenocorticism
Hypoadrenocorticism, caused by lack of mineralocorticoids and glucocorticoids, is most often diagnosed in young female dogs and usually has an immune-mediated etiology. Certain breeds, such as Leonbergers, standard poodles, and Portuguese water dogs, are at increased risk for the disease; however, hypoadrenocorticism may be diagnosed in any breed. Historical findings compatible with hypoadrenocorticism include intermittent vomiting, diarrhea, weight loss, Iethargyr, anorexia, and weakness. These symptoms often resolve with fluid therapy and corticosteroid treatment. Physical examination of animals in an acute hypoadrenal crisis reveals weak pulse, bradycardia, prolonged capillary refill time, severe mental slowness, and profound muscle weakness. Clinical features of hypoadrenocorticism that should raise suspicion include a normal or slow heart rate in the presence of circulatory shock and the “waxing and waning ’ course of disease before collapse.
Electrolyte abnormalities consisting of severe hyponatremia and hypochloremia associated with hyperkalemia are the hallmarks of hypoadrenocorticism. Azotemia and hyperphosphatemia also accompany primary hypoadrenocorticism, which makes it difficult to differentiate it from acute renal failure. Azotemia may be prerenal as a result of dehydration and hypovolemia, or increase in BUN may be caused by gastrointestinal hemorrhage. Hematological abnormalities consist of eosinophilia and lymphocytosis, or eosinophil and lymphocyte counts may be normal in the presence of severe metabolic stress. The anemia of hypoadrenocorticism has classically been attributed to lack Ofglucocorticoid effects on the bone marrow. However, more recent studies have suggested that hemorrhagic gastroenteritis contributes significantly to the anemia. Although hypoglycemia is more common with secondary or atypical hypoadrenocorticism, it is rarely seen with typical hypoadrenocorticism.
Urine specific gravity is frequently low, attributable to medullary washout (inadequate medullary gradient caused by sodium depletion) and decreased medullary blood flow. Dilute urine in the presence of azotemia and hyperkalemia may easily be mistaken for acute renal failure. I Iormonal assays are necessary to confirm the presence or absence of adrenal disease and to differentiate between hypoadrenocorticism and renal failure.
Diagnosis of primary hypoadrenocorticism is based on clinical signs, classic electrolyte imbalances, and confirmation with a corticotropin response test. The baseline cortisol sample should be collected with the initial blood work, and synthetic corticotropin (Cosyntropin ∣Cortrospi], 0.25 mg) should be administered intravenously during the initial fluid therapy. A l-hour, postcorticotropin sample may then be drawn, and glucocorticoids may be administered after the 1-hour sample is taken. Intramuscular injection of corticotropin (gel or synthetic) may not be absorbed in animals in circulatory shock; therefore, intravenous administration of synthetic corticotropin is preferred. If glucocorticoids must be administered before the measurement of cortisol, dexamethasone sodium phosphate is preferred because it does not interfere with the cortisol assay. Endogenous plasma corticotropin may be measured to determine whether the hypoadrenocorticism is primary or secondary.
Dogs and cats with primary hypoadrenocorticism exhibit a subnormal response to corticotropin administration. Both the baseline and the postcorticotropin cortisol concentrations are usually low or undetectable. Endogenous plasma cortico-
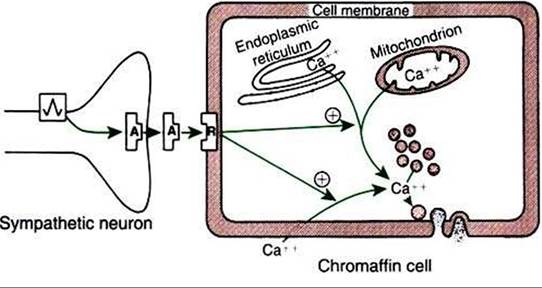
FIGURE 34-15 Stimulus-secretion coupling in the adrenal chromaffin cell. Note that cytosolic calcium may be derived from intracellular or extracellular sources. Circled plus signs indicate stimulation. A, Acetylcholine; Rz receptor. (From Hedge GAz Colby HDz Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
tropin concentrations are dramatically increased in animals with primary hypoadrenocorticism as a result of loss of negative feedback to the pituitary gland, caused by decreased serum cortisol concentrations. In the case of secondary hypoadrenocorticism, caused by a pituitary deficiency of corticotropin, the endogenous corticotropin concentrations are typically decreased (PK, protein kinase; PK-C, protein kinase C; PLC, phospholipase C. (From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
affect mainly the heart, and β2 receptors affect smooth muscle contraction and intermediary metabolism. Whereas all adrenergic receptors are responsive to both epinephrine and norepinephrine, the responses to the two catecholamines are different. In addition, the receptor types on various tissues vary in number, which, together with the different responses of adrenergic receptors on tissues, results in variable adrenergic responses being produced by a particular catecholamine.
The metabolic effects of catecholamines are mediated mainly by β2 receptors. Because epinephrine is 10 times more potent than norepinephrine with β2 receptors, epinephrine plays a much more important role in the control of intermediary metabolism than does norepinephrine. The effects of epinephrine on glucose metabolism are similar to those of glucagon and opposite to those of insulin. Epinephrine increases blood glucose concentrations, with the effect mainly in the liver; that is, epinephrine promotes both hepatic glycogenolysis and gluconeogenesis. Epinephrine also stimulates glycogenolysis in skeletal muscle, which in this situation is in contrast with the action of glucagon. Because glucose-6- phosphatase is not present in skeletal muscle, lactate is produced instead of glucose; the liver takes up lactate and converts it to glucose. Additional effects on glucose metabolism include the inhibition of insulin secretion (through α receptors) and stimulation of glucagon secretion by the pancreas; both actions increase blood glucose concentrations.
Epinephrine promotes lipolysis through interaction with two receptors on adipose cells. Activation of a lipase enzyme results in an increase in free fatty acids in the blood. Glucocorticoids potentiate the effect of epinephrine on lipolysis.
Catecholamines stimulate cardiac function. Both epinephrine and norepinephrine interact with β1 receptors to increase both the force of contraction and the heart rate, the latter resulting from the promotion of a shorter period of diastolic depolarization. Whereas both catecholamines promote arteriolar constriction through interaction with α receptors, epinephrine, through its high affinity for β2 receptors, causes the dilation of blood vessels both in the heart and in skeletal muscle. The end result is that total peripheral resistance is decreased by the action of epinephrine, with a concomitant decline in diastolic pressure; however, blood pressure is minimally changed, and cardiac output increases because of the increase in heart rate. The action of epinephrine to increase cardiac output is an obvious beneficial effect in situations that are described as “fight or flight.”
Catecholamines affect smooth muscle. Epinephrine causes relaxation of bronchial smooth muscle, particularly when the muscle is in a contracted state. Because the action is mediated through β2 receptors, norepinephrine has little effect on bronchial smooth muscle. Epinephrine causes relaxation of the smooth muscle of the gastrointestinal (Gl) tract through interaction with β2 receptors. Catecholamine stimulation of β-adrenergic receptors results in contraction of uterine smooth muscle, and stimulation of β2 receptors results in relaxation. Because of its dominant effect on β2 receptors, epinephrine causes relaxation of the uterus, whereas both epinephrine and norepinephrine interact with α receptors to cause contraction.
The effects of the catecholamines on bladder smooth muscle depend on different locations of α and β receptors; α-adrenergic receptors are located within the neck of the bladder, and β-adrenergic receptors are located within the body of the bladder. Epinephrine relaxes the body and contracts the neck of the bladder; norepinephrine contracts the neck of the bladder. The net effect is retention of urine.
Although the parasympathetic nervous system is the principal system involved in penile erection, the sympathetic nervous system may also play a role. Epinephrine promotes erection through vasodilation of the vasculature mediated by β receptors. Higher concentrations of epinephrine (and norepinephrine) can cause ejaculation through α-receptor interaction and vasoconstriction.
In the eye, epinephrine causes relaxation of the lens through stimulation of β receptors on the ciliary muscles. It causes dilation of the pupil through stimulation of α receptors, with resultant contraction of the radial muscle of the iris.
Table 34-5
Responses of Target Tissues to Catecholamines
| Target tissue | Receptor type | Responses |
| Liver | β2 | Glycogenolysis, lipolysis, gluconeogenesis |
| Adipose tissue | β2 | Lipolysis |
| Skeletal muscle | β2 | Glycogenolysis |
| Pancreas | α2 | Decreased insulin secretion |
| β2 | Increased insulin secretion | |
| Cardiovascular system | βl | Increased heart rate, increased contractility, increased conduction velocity |
| «2 | Vasoconstriction | |
| β2 | Vasodilation in skeletal muscle arterioles, coronary arteries, and all veins | |
| Bronchial muscle | β2 | Relaxation |
| Gastrointestinal tract | β2 | Decreased contractility |
| Urinary bladder | «2 | Sphincter contraction |
| β2 | Detrusor relaxation | |
| Uterus | a2 | Contraction |
| β2 | Relaxation | |
| Male sex organs | «2 | Ejaculation, detumescence |
| β2 | Erection? | |
| Eye | Radial muscle contraction | |
| β2 | Ciliary muscle relaxation | |
| Central nervous system | Ct2 | Stimulation |
| Skin | Ct2 | Piloerection, sweat production |
| Renin secretion | βl | Stimulation |
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
The effects of epinephrine on the CNS are excitatory. Drugs that affect the CNS probably do so by modulation of catecholamine concentrations» whereby sedation is associated with lower values of epinephrine. Other effects of catecholamine include the promotion of sweating and piloerection. Epinephrine also increases renin production by the renal juxtaglomerular cells. Table 34-5 summarizes the effects of catecholamines.