The pancreas has important endocrine and nonendocrine functions. The nonendocrine functions result from activity of the exocrine part of the pancreas and are involved in GI function.
The endocrine portion of the pancreas is organized as discrete islets (islets of Langerhans) that contain four cell types, each of which produces a different hormone (Figure 34-19).
The most numerous of the islet cells are β cells, which produce insulin; α cells produce glucagon; D cells produce somatostatin; and F or PF cells produce pancreatic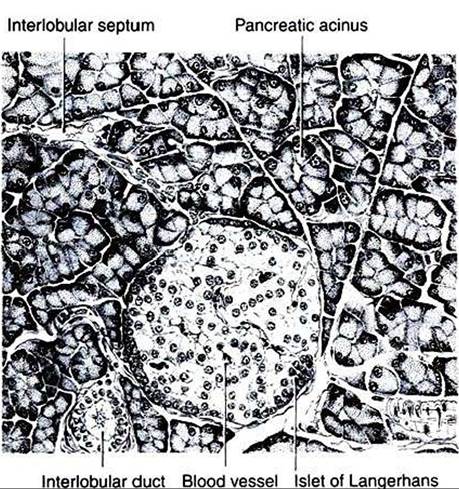
FIGURE 34-19 Depiction of section through the pancreas of the rat.The islet of Langerhans is a gland of internal secretion, whereas the surrounding acinar tissue forms an exocrine gland. (FromTurner CD, Bagnara JT: General endocrinology, ed 6, Philadelphia, 1976, Saunders.)
polypeptide (Figure 34-20). Although these hormones have different functions» all are involved in the control of metabolism and, more specifically, in glucose homeostasis.
Insulin
The first studies that associated the pancreas with carbohydrate metabolism were done by von Mering and Minkowski in 1889, when they showed that pancreatectomy of dogs resulted in signs similar to those characteristic of diabetes mellitus. Later, Banting and Best were able to show that injection of pancreatic extracts could alleviate the signs of diabetes mellitus in dogs and humans. Able was the first to crystallize insulin, and its structure was elucidated by Sanger in 1960.
Insulin is a protein consisting of two chains, designated A (21 amino acids) and B (30 amino acids), that are connected by two
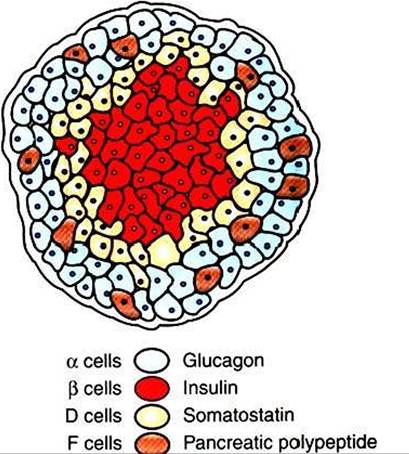
FIGURE 34-20 Depiction of the pancreatic islet. (From McDonald LE: Veterinary endocrinology and reproduction, ed 4, Philadelphia, 1989, Lea & Febiger.)
disulfide bridges. The monomer form of the hormone is thought to be the active form; insulin also exists in dimer and hexamer forms, the latter complexed with two zinc molecules.
Although there are some differences in amino acid composition among species, the differences are small; for example, cattle, sheep, horses, dogs, and whales differ only in positions 8,9, and 10 of the A chain. As a result, the biological activities of insulin are not highly species specific. Of the domestic species, feline insulin is most similar to bovine insulin, and canine insulin is identical to porcine insulin in its amino acid structure.The Synthesis of Insulin Is Biphasic: an Acute Phase Involves the Release of Preformed Insulin, and a Chronic Phase Involves the Synthesis of Protein
The synthesis of insulin, similar to that of other peptide hormones, begins with the formation of a linear polypeptide pre- proinsulin within the rough endoplasmic reticulum. A small peptide fragment is removed to form proinsulin. Proinsulin is coiled, and the end fragments are joined by disulfide bonds. Proinsulin is transferred to the GoIgi apparatus, where it is further processed and packaged into granules that contain both insulin and the connecting or C peptide (33 amino acids in length).
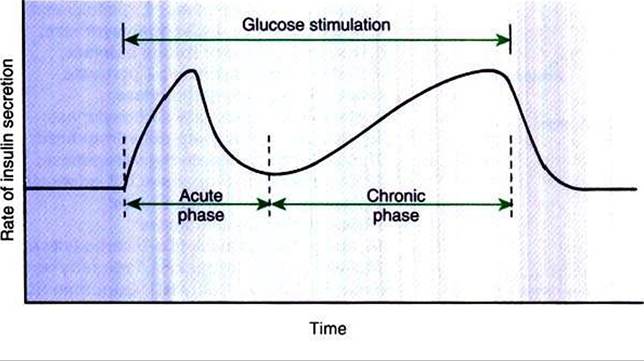
FIGURE 34-21 Kinetics of insulin secretion by the β cell in response to a continued glucose stimulus. (From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiofogy, Philadelphia, 1987, Saunders.)
The secretion of insulin follows biphasic kinetics in response to appropriate stimuli (Figure 34-21). The initial, acute release of insulin involves the exocytosis of preformed insulin from secretion granules. After the acute phase, a chronic phase of secretion occurs that involves the synthesis of protein and thus probably the synthesis of insulin.
The Metabolism of Insulin Involves Splitting the A and B Chains and Reducing the Chains to Amino Acids and Peptides
Insulin is metabolized mainly by the liver and kidneys. Enzymes that are present reduce the disulfide bonds that link the A and B chains, and the chains are then subjected to protease activity, which reduces them to peptides and amino acids.
The half-life of insulin is about 10 minutes.The Main Metabolic Functions of Insulin Are Anabolic
Insulin acts at a number of sites within the metabolic pathways of carbohydrates, fats, and proteins (Figure 34-22). It is important to realize that the liver is an especially important target organ, in part because the pancreatic venous effluent passes directly to the liver. The net effect of the actions of insulin is to lower blood concentrations of glucose, fatty acids, and amino acids and to promote intracellular conversion of
Table 34-6
Sites of Action and Effects of Insulin on Carbohydrate, Lipid, and Protein Metabolism
| Process affected | Site of action | ||
| Liver | Muscle | Adipose | |
| Carbohydrate metabolism | |||
| ↑Glucose transport | X | X | |
| TGIycogen synthesis | X | X | X |
| IGlycogenoIysis | X | X | X |
| TGluconeogenesis | X | ||
| Lipid metabolism | |||
| TLipogenesis | X | X | |
| TLipoIysis | X | X | |
| Protein metabolism | |||
| ↑Amino acid uptake | X | ||
| TProtein synthesis | X | ||
| TProtein degradation | X | ||
| TGluconeogenesis | X | ||
Frorr Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
these compounds to their storage forms: glycogen, triglycerides, and protein, respectively (Table 34-6). Glucose does not readily penetrate cell membranes except in a few tissues, such as brain, liver, and red and white blood cells, all of which must have continual access to glucose.
The presence of insulin is critical to the movement of glucose through the plasma membrane into the cell.360" class="lazyload" data-src="/files/uch_group31/uch_pgroup304/uch_uch7229/image/image357.jpg">
FIGURE 34-22 Metabolic pathways affected by insulin.The numbers correspond to each of the following enzymes:
T glucose-6-phosphatase; 2, glucokinase;
3, phosphorylase; 4, glycogen synthase;
5, fructose-1,6-bisphosphate aldolase;
6, 6-phosphofructokinase; 7, pyruvate kinase; 8, pyruvate carboxylase;
9, phosphoenolpyruvate carboxykinase;
10, glucose-6-phosphate-dehydrogenase;
11, 6-phosphogluconate dehydrogenase;
12, pyruvate dehydrogenase; 13, adenosine triphosphate (ATP)-Citrate lyase;
14,hormone-sensitive lipase;
15,acetyl-coenzyme A (CoA) carboxylase;
16, fatty-acid synthase. FFA, Free fatty acid. (From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Insulin has profound effects on carbohydrate metabolism. Insulin facilitates the use of glucose: namely, glycolysis, which involves the oxidation of glucose to pyruvate and lactate through the induction of enzymes, such as glucokinase, phosphoffuctokinase, and pyruvate kinase. Insulin promotes glycogen production in the liver, in adipose tissue, and in skeletal muscle by increasing glycogen synthetase activity with a concomitant decrease in glycogen phosphorylase activity. Gluconeogenesis is decreased by insulin because of the promotion of protein synthesis in peripheral tissues, thereby decreasing the amount of amino acids available for gluconeogenesis. In addition, insulin decreases the activities of hepatic enzymes (fructose 1,6-bisphosphate aldolase, pyruvate carboxylase, phosphoenolpyruvate carboxylase, and glucose-6- phosphatase) that are involved in the conversion of amino acids to glucose.
In adipose tissue, insulin promotes the synthesis of triglycerides. Insulin facilitates the intracellular use of glucose, which results in increased levels of pyruvate, a precursor of acetyl-coenzyme A (CoA) (in turn, a precursor of fatty acids) and increased glycerol 3-phosphate for the esterification of fatty acids.
Insulin activates the enzymes pyruvate dehydrogenase and acetyl-CoA carboxylase, which promote the synthesis of fatty acids from acetyl-CoA. Insulin also increases the activity of lipoprotein lipase located in the endothelium of capillaries of extrahepatic tissues, which promotes the movement of fatty acids into adipose tissue. Finally, insulin decreases lipolysis in adipose tissue.With protein metabolism, insulin promotes uptake of amino acids by most tissues, including skeletal muscle, but not liver. Insulin promotes protein synthesis and inhibits protein degradation. Therefore, insulin promotes the maintenance of a positive nitrogen balance. With insulin deficiency, protein catabolism increases, with increased amounts of amino acids available for hepatic gluconeogenesis and a resultant increase in the blood glucose concentrations.
The most important factor in the control of insulin secretion is the concentration of blood glucose. Increased concentrations of blood glucose initiate the synthesis and release of insulin by the β cells of the pancreatic islets (Figure 34-23). Two theories explain the mechanism of cellular induction of insulin synthesis and release. In the first, insulin exists within the plasma membrane, whereby glucose interacts with a membrane receptor protein that directs intracellular events toward the synthesis and release of insulin. In the second theory, insulin occurs at the intracellular level, whereby the metabolism of glucose produces the signal for insulin synthesis and release. Glucose control of insulin secretion is a positivefeedback system in which increased concentrations of glucose lead to increased concentrations of insulin.
Because the oral administration of glucose produces a greater insulin response than systemic administration, factors from the intestinal tract were thought to affect insulin secretion. It is now known that a number of GI hormones stimulate insulin secretion, including gastrin, cholecystokinin, secretin, and gastric inhibitory peptide.
The presence of amino acids and fatty acids in the intestinal tract also stimulates the release of insulin, although with less potency than that of glucose (Box 34-1).Hormones other than those from the Gl tract are important for the control of insulin secretion. Glucagon from the α cells of the pancreas has a direct stimulatory effect on the β cells to secrete insulin. Conversely, somatostatin inhibits
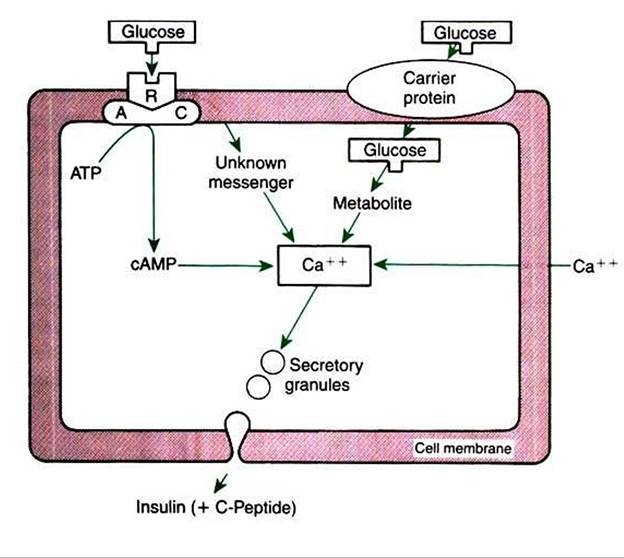
FIGURE 34-23 Proposed mechanisms of action of glucose on insulin secretion by the β cell. AC, Adenyl cyclase; ATP, adenosine triphosphate; R, receptor; cAMP, cyclic adenosine monophosphate. (From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Box 34-1
Factors Affecting Insulin Secretion
Stimuli
Glucose
Amino acids
Fatty acids
Gastrin
Pancreozymin-cholecystokinin
Secretin
Gastric inhibitory polypeptide
Glucagon Acetylcholine
Inhibitors
Somatostatin Epinephrine Norepinephrine
From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.
the secretion of insulin. Both hormones work through the adenyl cyclase system, glucagon being stimulatory and somatostatin being inhibitory. CatechoIamines tend to decrease insulin secretion through an interaction with the α-adrenergic receptors on the β cells. Whereas epinephrine is the main circulating catecholamine that affects insulin secretion, norepinephrine also influences insulin secretion because the pancreas has adrenergic innervation by the autonomic nervous system. The pancreas also has cholinergic innervation by the autonomic nervous system, and in contrast to adrenergic stimulation, cholinergic activity increases insulin secretion through the release of acetylcholine.
Insulin Deficiency Produces Diabetes Mellitus, Which Can Culminate in Diabetic Ketoacidosis
A lack or deficiency of insulin produces a syndrome called diabetes mellitus (DM). DM may be type /, which is most common in dogs, or type 2, which is most common in cats. Type 1 DM is caused by an autoimmune destruction of the β cells of the pancreas; it leads to absolute insulin deficiency and a propensity to develop ketosis. Diabetic ketoacidosis (DKA) is the culmination of DM that results in unrestrained ketone body formation in the liver, metabolic acidosis, severe dehydration, shock, and possibly death. Hepatic lipid metabolism becomes deranged with insulin deficiency, and non- esterified fatty acids are converted to acetyl-CoA rather than incorporated into triglycerides. Acetyl-CoA accumulates in the liver and is converted into acetoacetyl-CoA, then ultimately to ketones, including acetoacetic acid, β-hydroxybutyrate (primary ketone in dogs and cats), and acetone.
As insulin deficiency culminates in DKA, accumulation of ketones and lactic acid in the blood and loss of electrolytes and water in the urine result in profound dehydration, hypovolemia, metabolic acidosis, and shock. Ketonuria and osmotic diuresis, resulting from glycosuria, cause sodium and potassium loss in the urine, exacerbating hypovolemia and dehydration. Nausea, anorexia, and vomiting, caused by stimulation of the chemoreceptor trigger zone through ketonemia and hyperglycemia, contribute to the dehydration caused by osmotic diuresis. Dehydration leads to further accumulation of glucose and ketones in the blood. Stress hormones such as cortisol and epinephrine contribute to the hyperglycemia in a vicious cycle. Eventually, severe dehydration may result in hyperviscosity, thromboembolism, severe metabolic acidosis, renal failure, and finally death.
Historical Findings. Most dogs and cats with DKA present with a previous history of uncomplicated diabetes, including polyuria, polydipsia, and dramatic rapid weight loss in the presence of a good or even ravenous appetite. More recent historical findings include anorexia, weakness, depression, vomiting, and diarrhea. Occasionally, owners fail to notice the significance of the classic signs of DM, and the animals are presented solely with an acute history of DKA. DKA may also develop in previously well-controlled, treated diabetic patients.
Physical Examination Findings. The most common examination findings in DKA are lethargy and depression, dehydration, unkempt hair coat, and muscle wasting. Hepatomegaly is common in both diabetic cats and diabetic dogs. Cataracts are often observed in diabetic dogs. A plantigrade rear limb stance resulting from diabetic neuropathy is often observed in diabetic cats. Other findings include tachypnea, dehydration, weakness, vomiting, and occasionally a strong acetone breath odor. Cats may present recumbent or comatose, which may be a manifestation of mixed ketotic- hyperosmolar syndrome. Icterus can develop from the complicating factors of hemolysis, hepatic lipidosis, and acute pancreatitis.
Laboratory Findings. Cilucose level is greatly elevated. The average blood glucose concentration in patients with DKA is 25 mmol/L. Values can range from 10 mmol/L to greater than 50 mmol/L, but the latter is more characteristic of hyperosmolar coma. Although portable glucose meters are typically used to monitor glucose concentrations in DKA, caution is advised in relying on these monitors for baseline glucose concentrations because of inaccuracies in animals with severe hyperglycemia. All DKA patients have a relative or absolute deficiency of insulin and excessive hepatic production of glucose, resulting in hyperglycemia. Hyperglycemia is further exacerbated by dehydration and the corresponding reduction in GFR, and these factors are important determinants of its severity. This is supported by the findings that (1) glucose concentrations should exceed 25 mmol/L only when dehydration is severe enough to reduce GFR, and thus the ability of the kidneys to excrete glucose, and (2) fluid administration alone can significantly reduce blood glucose concentrations.
Osmolality usually is mildly to extremely increased in the DKA patient as a result of hyperglycemia, but (his may not be detected, in part because of concurrent hyponatremia. Sodium (and to a lesser extent potassium)» glucose, and urea concentrations are the determinants of the calculated serum osmolality. Reference values for serum osmolality in dogs and cats are approximately 290 to 310 mθsm∕kg. Hyperosmolality is generally mild enough to resolve with intravenous fluid and insulin therapies.
Nonketotic hyperosmolar diabetes is defined by extreme hyperglycemia (>30 mmol/L), hyperosmolality (>350 mOsm∕L), severe dehydration, and CNS depression, without ketone formation and with absent or mild metabolic acidosis. Affected patients are more likely to have underlying renal or cardiovascular disease and to be non-insulin dependent. Although this specific syndrome, as defined in humans, is infrequently encountered in veterinary medicine, ketotic or nonketotic diabetic cats may be seen, with significant hyperosmolality and CNS alterations.
Most patients with DKA have a total body K^ deficit caused by urinary (osmotic diuresis), anorexia, and Gl losses (vomiting, anorexia). The metabolic acidosis, relative or absolute insulin deficiency, and serum hypertonicity combine to cause a K+ shift from the intracellular to the extracellular compartment. This is capable of masking the severity of total-body hypokalemia when plasma concentrations are measured. Insulin therapy, as well as correction of the acid-base disturbance with fluids and bicarbonate, will drive serum K4 intracellularly, potentially causing marked circulating hypokalemia. Polyuric patients are predisposed to severe hypokalemia, whereas oliguric or anuric patients are predisposed to severe hyperkalemia.
In general, DKA causes significant total-body Na‘ deficits. Excessive urinary loss of Na+ results from the osmotic diuresis induced by high glucose and ketone concentrations and the lack of insulin, which usually aids in reabsorption of Na' from the distal nephron. HypergIucagonemia, vomiting, and diarrhea also contribute to the total-body Na* loss. Hyperosmolality may contribute to a low Na* concentration because as osmolality increases, water is drawn from the interstitium into the vascular space, thus diluting plasma Na* and CΓ.
Phosphorus is the major intracellular anion and is important for energy production and for maintenance of cell membranes. Concentrations are regulated by dietary intake, renal elimination, factors promoting its movement into and out of cells, and vitamin D and parathyroid interactions. In DKA, circulating concentrations are usually within reference range or increased initially because of dehydration and renal disease. Phosphorus may also be low at presentation because of urinary loss from osmotic diuresis. As long as renal function is not compromised, a significant decrease in phosphorus should be anticipated with therapy. After insulin administration, phosphorus shifts to the intracellular compartment with glucose. Clinical signs of hypophosphatemia, such as hemolytic anemia (also seen with Heinz bodies in DKA), lethargy, depression, and diarrhea, may develop once concentrations reach 0.32 mmol/L. OversuppIementation of phosphorus should be avoided because hypocalcemia or metastatic calcification may result.
Magnesium (total serum) is not usually measured routinely, but concentrations may be abnormal in DKA. A recent study in cats demonstrated high total serum magnesium concentrations at presentation in those with DKA; after 48 hours of therapy, however, total serum magnesium concentrations were significantly decreased. Magnesium deficiency may be caused by poor oral intake, decreased intestinal absorption, increased renal loss, or changes in distribution because it is the second most abundant intracellular cation. Clinical signs of hypomagnesemia include neuromuscular weakness and cardiac arrhythmias, signs that can be seen with other electrolyte alterations. Hypomagnesemia can also cause decreases in other electrolytes, such as potassium and calcium. Correction of deficits may resolve electrolyte disturbances and may improve clinical outcome in the severely deficient patient.
Liver enzyme elevations are common in DM patients. Further increases potentially occur in DKA. Alanine aminotransferase and aspartate aminotransferase are most often affected, increasing secondary to hypovolemia and poor hepatic blood flow, with subsequent hepatocellular damage. Further increases in serum alkaline phosphatase concentration may occur if pancreatitis and secondary cholestasis ensue. Cholesterol and triglycerides may be elevated secondary to derangements of lipid metabolism resulting from decreased insulin.
Metabolic acidosis is one of the most prominent features of DKA. As ketone bodies accumulate in the blood and overwhelm the body’s buffering capabilities, there is an increase in hydrogen ions and a decrease in bicarbonate. As dehydration worsens, blood flow to peripheral tissues decreases, and the resulting lactic acidosis may contribute to the acid-base disturbance. Acidosis may manifest as lethargy, vomiting, hyperventilation, decreased myocardial contractility, peripheral vasodilation, stupor, and coma. Initiation of insulin therapy (to stop ketogenesis) and fluid therapy (to correct dehydration) will help improve the metabolic acidosis in most patients. Bicarbonate supplementation should be pursued with caution and is generally not recommended unless the patient’s blood pH is less than 7.1 or serum bicarbonate is less than 12 mmol/L.
Anion gap may be normal or elevated. An elevated value further characterizes the metabolic acidosis caused by DKA. The anion gap is a representation of the circulating anions not routinely measured on biochemical analyses. The normal anion gap ranges from 10 to 20 and is calculated by the following equation:
Osmolality (mθsm) = 2(Na4 + K4 [mEq∕L]) + Glucose (mmol/L)
+ BUN (mmol/L)
In DKA the ketones become unmeasured anions as they dissociate from ketoacids. However, if significant dehydration is present secondary to the osmotic diuresis and vomiting, lactic acidosis secondary to tissue hypoxia may contribute to the unmeasured anions, thus increasing the anion gap.
Circulating urea and creatinine concentrations may be within reference range or high. These values are high in most patients because of severe dehydration, but renal insufficiency or failure is also a possible cause. Increases in urea and creatinine must be evaluated in light of the urine specific gravity. A low urine specific gravity at presentation does not always guarantee a diagnosis of renal insufficiency or failure, because osmotic diuresis and chronic hypokalemia can contribute to low specific gravities in DM patients. Therefore, reevaluation of urea, creatinine, and urine specific gravity must be done after treatment of the crisis. If urea and creatinine are initially elevated and remain static or increase with appropriate therapy, concurrent renal disease is strongly suspected.
The most important part of urinalysis is measurement of glucose and ketones. A strongly positive glucose result confirms DM, and a positive result for ketones confirms DKA. However, a negative ketone result does not definitively rule out ketosis. The nitroprusside reagent used in urine sticks detects only acetoacetate and acetone. It is not as sensitive for β-hydroxy- butyrate, the most prevalent ketone body, and therefore may be negative in the presence of ketosis. A recent study reported that β-hydroxybutyrate concentrations greater than 1.9 mmol/L were the most sensitive indicator of DKA, and values greater than 4.8 mmol/L were highly specific for its diagnosis. Using a cutoff value of 3.8 mmol/L was associated with the best combination of specificity (95%) and sensitivity (72%) for DKA.
The presence of pyuria and hematuria on urinalysis, along with confirmation by examination of urine sediment, supports the presence of UTL However, urine culture should be performed regardless of urine sediment activity.
The hemogram may be normal at presentation but usually reveals a leukocytosis with a mature neutrophilia (common in cats) or a stress Ieukogram. There may be a regenerative or degenerative left shift suggestive of a severe inflammatory and infectious process. The red blood cell count and hematocrit may be elevated as a result of dehydration. Heinz bodies, with or without anemia, may be noted in cats because feline hemoglobin is uniquely susceptible to oxidative damage.
Concurrent Disease. Frequently, an underlying stressful event precipitates the shift from DM to DKA or nonketotic hyperosmolar DM. Impaired immune function secondary to DM increases the risk of infections. The precipitating event may be a UTI or other viral or bacterial infection or inflammatory disorder, such as pancreatitis, pyelonephritis, cholangio- hepatitis, inflammatory bowel disease (IBD), eosinophilic granuloma complex, prostatitis, pyometra, upper respiratory infection, or pneumonia. Other concurrent diseases may include renal insufficiency or failure, hepatic lipidosis, neoplasia, and congestive heart failure. Recent drug therapy may also precipitate a crisis, especially administration of corticosteroids or progestagens. Therefore, further diagnostic testing of the diabetic patient that presents in a crisis is essential, particularly abdominal radiography or ultrasonography, as well as thoracic radiography and echocardiography if indicated. Additional testing for concurrent endocrine diseases, such as hyperthyroidism in cats and hypothyroidism and Fiyperadrenocorticism in dogs, may also be indicated but should be postponed until some control of the DM is achieved, because uncontrolled disease may affect the results of the tests.
Ancillary tests for pancreatitis include abdominocentesis or diagnostic peritoneal lavage if pancreatitis is suspected. Serum amylase and lipase, if determined on presentation, may be elevated in the absence of pancreatitis, secondary to severe dehydration or renal insufficiency, and demonstration of an elevated circulating concentration of trypsin-like immunoreactivity (TLI) may be preferable. Cats and dogs with acute necrotizing pancreatitis usually present with vomiting, abdominal pain, and concurrent DKA. Physical examination findings include icterus, cranial abdominal pain, and abdominal effusion. Radiographs may reveal a “ground glass” appearance of the abdomen, and abdominal ultrasound usually shows enlargement and hypoechogenicity of the pancreas. Concurrent hepatopathies are often present in patients with DKA, but evaluation is complicated by the effect of both DM and DKA on liver enzymes and liver function tests. Ultrasonography may be more useful in these cases. Diagnostic peritoneal lavage is usually necessary to demonstrate inflammatory, nonseptic peritonitis, and abdominal lipase is usually increased dramatically in affected cats and dogs.
Dietary Management Is an Important Consideration in Therapy for FeIineType 2 Diabetes
Type 2 DM is caused by insulin resistance and secondary β-cell failure. Type 2 diabetes may be managed using oral hypoglycemic agents, diet, or insulin. DM is one of the most common feline endocrinopathies, affecting 1 in 300 cats. The pathogenesis of type 2 DM in cats is reviewed earlier. Diagnosis of DM can be challenging, particularly in the early stages when the cats are non-insulin dependent. However, once clinical signs of diabetes are observed (polyuria, polydipsia, neuropathy), many cats may still benefit from alternatives to insulin therapy. In general, the primary abnormalities associated with type 2 DM, such as obesity and insulin resistance, are reversible. Insulin secretory ability, however, may be reversible (glucose toxicity) or irreversible (pancreatic amyloid deposition). In cats the differentiation of insulin-dependent (type 1) DM and non-insulin-dependent (type 2) DM is virtually impossible before treatment; therefore the clinician may have to rely on the response to oral hypoglycemic agents as a guide to whether the cat has sufficient β-cell function to be managed with oral hypoglycemic agents.
Goals of therapy for DM include restoration of normal fasting serum glucose concentrations, normalization of serum fructosamine, and reversal or attenuation of chronic complications, such as diabetic neuropathy and nephropathy. As in human patients with type 2 DM, the best approach to cats is a stepwise progression from dietary management to oral hypo- glycemics and finally to insulin therapy when “islet burnout” occurs.
Exercise and diet is the Cornerslone of therapy in human patients with type 2 DM. In most diabetic cats, exercise is not a reasonable option. One mechanism by which cats may be encouraged to exercise is by feeding the cat multiple small meals hidden in various places within the house. For example, an obese diabetic cat might be encouraged to jump up on the refrigerator or counter to find small amounts of food and then have to hunt for the rest of the food at the opposite end of the house.
In human diabetic patients, fiber supplementation is beneficial in the management of the disease. In humans and dogs, increased amounts of fiber slow the rate of glucose absorption from the intestine and minimize the postprandial fluctuations in blood glucose. This allows better glycemic control and correction of obesity; however, the data in cats are less compelling. In the only study of high-fiber diets in cats, 9 of 13 diabetic cats showed significant improvement in glycemic control with consumption of a high-fiber diet. Examples of high-fiber diets include prescription diet w/d and r∕d, Science Diet Maintenance Light, Purina OM, and lams Less Active. Because many cats find high-fiber diets unpalatable, soluble fiber such as psyllium can be mixed into the cat’s regular food, and glycemic control may still be enhanced. If the cat’s weight is normal at the start of therapy, the diet should be fed at a maintenance level of 60 to 70 kcal/kg/day. If the patient is obese, caloric intake should be limited to 70% to 75% of the energy needs for the cats optimal weight.
The cat is an obligate carnivore and as such is unique among mammals in its insulin response to dietary carbohydrates, protein, and fat. The feline liver exhibits normal hexo- kinase activity, but glucokinase activity is virtually absent. Glucokinase converts glucose to glycogen for storage in the liver and is important in “mopping up” excess postprandial glucose. Normal cats are similar to diabetic humans because glucokinase levels drop precipitously with persistent hyperglycemia in humans with type 2 DM. Amino acids, rather than glucose, are the signal for insulin release in cats. In fact, a recent study demonstrated more effective assessment of insulin reserve in cats using the arginine response test rather than a glucose tolerance test. Another unusual aspect of feline metabolism is the increase in hepatic gluconeogenesis seen after a normal meal. Normal cats maintain essential glucose requirements from gluconeogenic precursors (i.e., amino acids) rather than from dietary carbohydrates. As a result, cats can maintain normal blood glucose concentrations even when deprived of food for over 72 hours; furthermore, feeding has negligible effect on blood glucose concentrations in normal cats. In summary, the cat is uniquely adapted to a carnivorous diet (mice) and is not metabolically adapted to ingestion of excess carbohydrate.
When type 2 DM occurs in cats, the metabolic adaptations to a carnivorous diet become even more deleterious, leading to severe protein catabolism; feeding a diet rich in carbohydrates may exacerbate hyperglycemia and protein wasting in these diabetic cats. In humans with type 2 diabetes, the first recommendation is to restrict excess dietary carbohydrates such as potatoes and bread and to control obesity by caloric restriction. Furthermore, human patients with type 2 DM have improved glycemic control and nitrogen turnover during weight loss when a low-energy diet (high protein) is combined with oral hypoglycemic therapy.
The authors have found high-protein diets to he beneficial in increasing lean body mass and reducing postprandial hyperglycemia. Caution should be used when high-protein, restricted- Carbohydrate diets are used in cats also treated with insulin because the insulin requirement may decrease. Usually the insulin dose is decreased by 25% in cats given high-protein diets and insulin. On the other hand, high-protein diets and oral hypoglycemic agents appear to be complementary treatments in cats that are underweight.
Glucagon
GIucagon is a protein hormone produced by the α cells of the islets of Langerhans. It has a close relationship with insulin in the control of glucose metabolism.
Glucagon is a polypeptide consisting of a single chain composed of 29 amino acids. There is considerable homology in amino acid composition among species. Glucagon is produced in other sites besides the pancreas; the stomach produces a molecule called gut glucagon that is identical to the pancreatic glucagon molecule, and the small intestine produces an immunologically similar molecule called glicentin. As with other polypeptide hormones, glucagon is first synthesized in the endoplasmic reticulum as part of a precursor molecule, packaged in the Golgi apparatus, and final processing occurs in the secretory granules. Glucagon is released by exocytosis. Glucagon is metabolized mainly by the liver and kidneys. It has a half-life in plasma of about 5 minutes.
The Most Important Functions of Glucagon Are to Decrease Glycogen Synthesis, Increase Glycogenolysis, and Increase Gluconeogenesis
The physiological actions of glucagon are the opposite of those of insulin; the main effect of glucagon is centered in the liver. Glucagon increases cAMP production in the liver, which leads to decreased glycogen synthesis, increased glycogenolysis, and increased gluconeogenesis, the last related to the effects of glucagon on protein metabolism (Figure 34-24). The net result is an increase in glucose concentrations in the blood.
Changes in glucagon secretion counterbalance the effects of insulin in association with the daily ingestion of food and the intervals between food intake periods. After the consumption of food, the initial response of the metabolic system is increased insulin secretion, which results in conservation of energy through the formation of storage forms of carbohydrates, lats, and proteins. Glucagon secretion, which begins with the ingestion of food, increases as the interval from food ingestion lengthens and blood glucose concentrations begin to decline. This secretion allows the individual to mobilize energy stores for the maintenance of glucose homeostasis (i.e., to prevent postprandial hypoglycemia) (Figure 34-25).
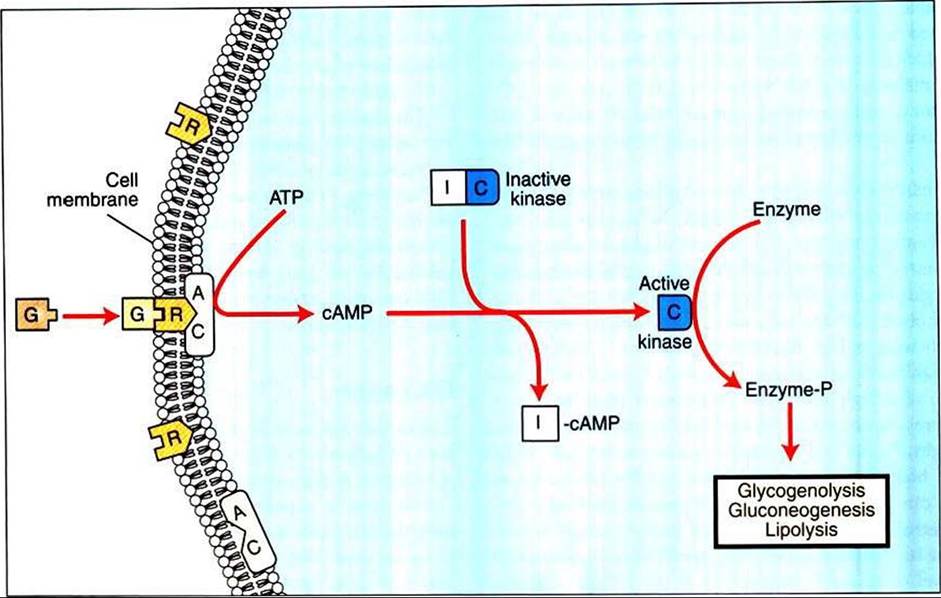
FIGURE 34-24 Mechanism of action of glucagon ∣G) on its target cells. AC, Adenyl cyclase; ATP, adenosine triphosphate; cAMP cyclic adenosine monophosphate; / and C, inhibitory and catalytic subunits of the kinase, respectively; P, receptor. (From Hedge GA, Colby HD, Goodman RL: Clinical endocrine physiology, Philadelphia, 1987,
Saunders.)
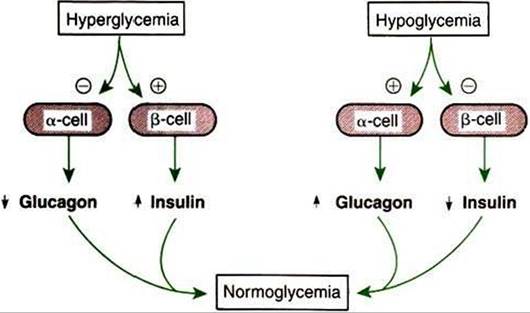
FIGURE 34-25 Effects of hyperglycemia and hypoglycemia on the secretion of insulin and glucagon by the pancreatic β cells and α cells, respectively. Plus signs indicate stimulation; minus signs indicate inhibition. (From Hedge GA, Colby HD, Good man RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Glucagon Synthesis Is Stimulated by Decreased Glucose Concentrations in the Blood
The main factor that regulates glucagon secretion is plasma glucose concentration. In contrast to insulin synthesis, decreased glucose concentrations stimulate glucagon synthesis and release, a relationship that represents a negative-feedback system. It must be remembered that glucagon regulation works in tandem with insulin regulation to maintain glucose concentrations within the physiological range. In fact, if glucagon were not secreted to maintain blood glucose concentrations, the individual would die of hypoglycemic shock. Because the α cells require insulin for glucose entry into the cells (as do most cells), in clinical syndromes involving insulin insufficiency (diabetes mellitus), glucose entry into the α cells is reduced, and plasma glucagon concentrations are paradoxically elevated. Glucagon promotes Iipolysis and an increase in fatty acids, which has a negative-feedback effect on glucagon secretion.
Protein ingestion represents an exception to the rule of opposite responses of glucagon and insulin. The release of both insulin and glucagon in response to protein ingestion appears logical; increased insulin secretion, in response to increased plasma amino acid levels, leads to lower glucose concentrations, and increased glucagon would counteract this through increased hepatic gluconeogenesis, resulting in maintenance of blood glucose within normal limits. The complementary responses of insulin and glucagon allow growth to occur in animals fed a diet only of protein and fat.
Intestinal hormones, with the exception of secretin, stimulate both glucagon and insulin secretion. A similar (inhibitory) response to somatostatin is observed for both glucagon and insulin. Both sympathetic and parasympathetic stimulation of the autonomic nervous system induce secretion of glucagon (Figure 34-26).
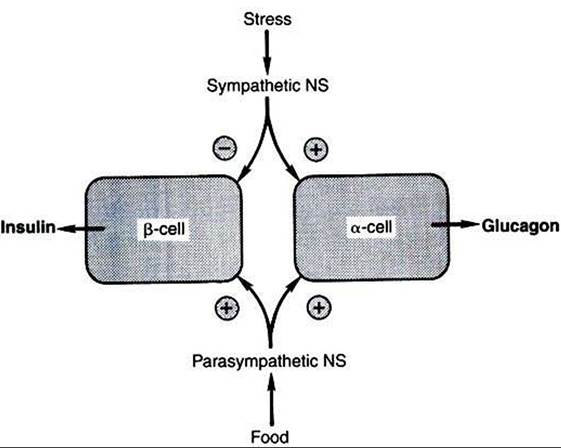
FIGURE 34-26 Regulation of insulin and glucagon secretion by the autonomic nervous system. Plus signs indicate stimulation; minus sign indicates inhibition. NS, Nervous system. (From Hedge GA, Colby HD, Good man RL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Some birds have a predominance of glucagon in the pancreas, which suggests that glucagon may have a more important role in carbohydrate metabolism of avian species than in mammals.
Somatostatin
As indicated in Chapter 33, somatostatin was first described in the brain as a 14-amino acid peptide that inhibits growth hormone secretion by the pars distalis. The molecule has since been identified in a number of tissues, including other areas of the brain, the GI tract, and the D cells of the pancreatic islets. Its synthesis and secretion are similar to those observed for other protein hormones. The metabolism of somatostatin is rapid (about 5 minutes) and occurs mainly in the liver and kidneys.
The Main Functions of Somatostatin Are to Inhibit the Secretion of Hormones Produced by the Pancreas (Insulin, Glucagon, Pancreatic Polypeptide)
The actions of somatostatin can be classified as inhibitory. Pancreatic somatostatin inhibits the digestive processes by decreasing nutritive absorption and digestion. The motility and secretory activity of the GI tract are decreased by somatostatin. One of the most important physiological functions of pancreatic somatostatin is the regulation of the endocrine cells of the pancreas (Figure 34-27). Somatostatin inhibits secretion of all endocrine cell types of the islets of Langerhans, including the D cells. The α cells are more affected by the inhibitory action of somatostatin than are β cells; therefore, glucagon secretion is more affected by somatostatin than is insulin secretion.
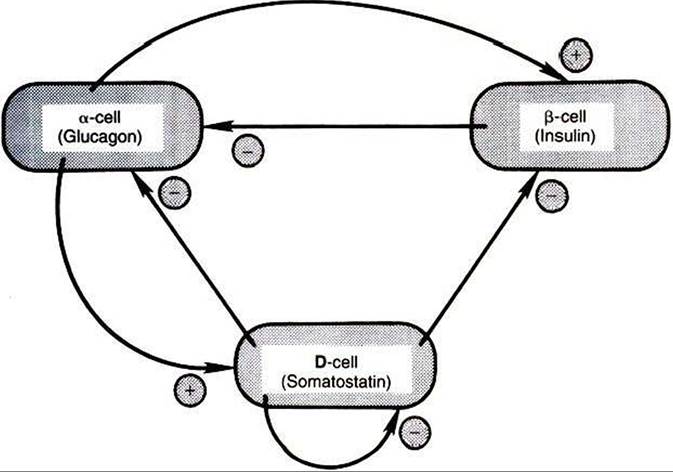
FIGURE 34-27 Possible cell-to-cell interactions in the pancreatic islets. Plus signs indicate stimulation; minus signs indicate inhibition. (From Hedge GA, Colby HD, Goodman HL: Clinical endocrine physiology, Philadelphia, 1987, Saunders.)
Somatostatin secretion is increased by nutrients (e.g., glucose» amino acids) and by the neurotransmitters of the autonomic nervous system (epinephrine, norepinephrine, acetylcholine). Of the hormones produced by the pancreas, only glucagon stimulates somatostatin secretion.
Pancreatic Polypeptide
Pancreatic polypeptide, a 36-amino acid polypeptide, is produced by the F cells of the pancreas (see Figure 34-20). In contrast to somatostatin secretion, pancreatic polypeptide secretion is limited to the pancreas.
The effects of pancreatic polypeptide are directed toward the GI tract. The secretion of pancreatic enzymes and the contraction of the gallbladder are inhibited by the actions of this hormone. Both gut motility and gastric emptying are increased by the action of pancreatic polypeptide.
The secretion of pancreatic polypeptide is stimulated by intestinal hormones, including cholecystokinin, secretin, and gastrin. Stimulation of the vagus nerve also promotes pancreatic polypeptide secretion. The ingestion of protein is stimulatory for secretion, whereas carbohydrates and fats have little effect. As indicated previously, somatostatin inhibits pancreatic polypeptide secretion.