Complications of liver disease
7.4.1 Ascites
Ascites is abdominal fluid of low protein content and low cel- lularity, such as can be found in patients with liver disease. Ascites occurs relatively frequently in dogs with chronic liver disease, but is rare in cats.
Intrahepatic portal venous hypertension is the most common cause leading to ascites in dogs.37,38 These patients also commonly show a reduced serum albumin concentration, so that decreased oncotic pressure and portal hypertension both contribute to the formation of ascites.39 In patients with pre-hepatic causes, such as an arteriovenous fistula or a portal vein thrombosis, the portal blood pressure is usually much higher and the albumin concentration does not decrease as much as it does with intrahepatic causes of ascites, such as chronic hepatitis. Portal vein occlusion may be caused by compression due to masses or a congenital portal vein hypoplasia. Postsinusoidal sphincters have been identified in hepatic veins of dogs and may add to venous outflow resistance. Also, sodium and water retention due to hyperaldosteronism may further enhance the formation of ascites. Extravasation of bile from a ruptured biliary tract or from the gall bladder elicits a strong inflammatory peritonitis with transudation of lymph. In such cases, the peritoneal fluid appears characteristically dark brown or green, and the inflammation causes the fluid to be turbid. Cytological evaluation of this fluid will show that it contains many neutrophils.40 Bile duct rupture may cause septic peritonitis due to a secondary infection, usually with anaerobic bacteria.7.4.2 Jaundice
Bilirubin is a waste product ofheme degradation (Figure 7.4).41 Heme mainly stems from the hemoglobin in erythrocytes (65% of all heme production), with a smaller contribution from myoglobin and heme-containing enzyme systems in the liver (30-35%).
Bilirubin produced from heme is cleared from the plasma, conjugated by the liver, and excreted into the bile. In the intestinal tract, conjugated bilirubin undergoes bacterial deconjugation and becomes reduced to urobilinogen. Uro-bilinogen is reabsorbed and then once again cleared by the liver (i.e., enterohepatic circulation). Only a very small amount of it escapes into the systemic circulation and is excreted in the urine. In the colon, urobilinogen is transformed into stercobilin, which gives the feces their normal brown color.
Hepatic handling (i.e., clearance, conjugation, and especially biliary excretion) of bilirubin may become impaired due to hepatic parenchymal and/or biliary diseases. Obstruction of the common bile duct near the duodenum results in impairment of all proximal mechanisms of bilirubin handling (i.e., excretion, conjugation, and clearance), but the resulting mixed conjugated/unconjugated hyperbilirubinemia consists predominantly of the conjugated or direct form. Cholestasis is also associated with increased serum activities of GGT and AP, and with increased serum bile acids concentrations. Biliary tract rupture allows the leakage of bile into the peritoneal cavity. The peritoneum then absorbs the pigments, which in turn leads to severe icterus. Severe hemolysis leads to increased bilirubin production. In addition, the liver develops centrolo- bular necrosis due to hypoxia. Icterus due to hemolysis is only seen when the hemolysis is acute and severe, resulting in mixed conjugated /unconjugated hyperbilirubinemia due to the combination of hypoxic necrosis and cholestasis with increased production of bilirubin.
The reference ranges for serum total bilirubin concentration in dogs and cats may vary from laboratory to laboratory, but most laboratories agree that concentrations above 0.3 mg/dL in cats and 0.6 mg/dL in dogs are abnormal.
Dogs, especially males, have the necessary renal enzyme systems to produce and conjugate bilirubin, so bilirubinuria may be a normal finding in urine samples from dogs.
In contrast, bilirubinuria is an abnormal finding in cats, is associated with hyperbilirubinemia, and is always pathological.Acholic feces may result from a total absence of pigment in the intestinal tract. Only a small amount of bile pigment is necessary to stain feces to their normal color. Complete EBDO due to cholelithiasis, a tumor of the bile duct, the head of the pancreas, or the duodenal wall may cause such acholic, greasy, gray-colored feces.
Unpigmented, concentrated mucoid bile may develop in patients with chronic severe extrahepatic bile duct obstruction. This “white bile syndrome,” may occur in both dogs and cats.
Conjugated bilirubin may easily and irreversibly (covalently) bind to albumin in the circulation.42 This permanently bound bilirubin is no longer available for normal hepatic clearance from the plasma and therefore stays in the circulation and other tissues until the albumin is degraded. As a consequence, animals may remain icteric for several weeks after the resolution of the cause of the icterus and icteric mucous membranes may not accurately reflect the present situation, but rather be representative of a previous disease state.
7.3.6 Hepatic encephalopathy
Hepatic encephalopathy (HE) is defined as a dysfunction of the brain secondary to hepatic dysfunction. HE occurs frequently in both dogs and cats with severe hepatic dysfunction and represents a complex of neurological symptoms.43,44 Like icterus, HE is not a diagnosis, as the causes of HE may be quite varied. HE can occur due to two different clinical scenarios, an acute severe total hepatic failure (also called fulminant hepatic failure) or a chronic form, which can be subclinical to severe.
Fulminant hepatic failure can be due to an acute and complete necrosis of the liver and may be caused by infections, such as canine adenovirus 1 or toxins, such as acetaminophen, fungal toxins (e.g., aflatoxicosis), and mushroom toxins (e.g., phal- loidin).
Fulminant hepatic failure causes severe HE or even hepatic coma. It also causes severe icterus, vomiting, and spontaneous bleeding tendencies due to DIC. The activities of liver enzymes in the serum or plasma are extremely elevated, and many such patients die within a few days. Fortunately, chronic HE is by far the most common form. Chronic HE can be caused by the shunting of portal blood so it bypasses the liver through the portosystemic collateral circulation. Portosystemic shunting may be inherited or acquired, with the latter form being caused by portal hypertension.In cats, there is another form of HE, which is related to a deficiency of essential amino acids and the development of hepatic lipidosis (see 7.6.1.1). This is the only form of HE that requires amino acid supplementation for successful management; all other forms of HE should be treated by a reduction of dietary protein. Patients with HE may also benefit from the administration of lactulose orally or by enema, in addition to antibiotic therapy with neomycin or another broad-spectrum antibiotic.
In both dogs and cats, chronic HE is most commonly caused by the shunting of portal blood past the liver and is thus also referred to as portosystemic encephalopathy. The great functional reserve of the liver protects animals against HE, even if they have severe liver disease. Even severe portosystemic shunting alone is not sufficient to cause HE in most cases and only leads to HE when present in combination with compromised hepatic function. This situation occurs, for example, in patients with chronic hepatitis leading to portal hypertension and acquired portosystemic collaterals. In dogs and cats with congenital shunts, the hepatic function also becomes increasingly inadequate. Normally, the liver grows due to the expression of
Table 7.2: Neurological symptoms in dogs and cats with hepatic encephalopathy (HE)
Neurological symptoms seen in dogs and cats with HE
Stage 1 Apathy, decreased mental alertness, “staring”, unawareness of surroundings
Stage 2 Ataxia, circling, head pressing against obstacles, blindness, salivation
Stage 3 Stupor, severe salivation, completely inactive but arouseable
Stage 4 Coma, totally unresponsive
Non-neurological signs associated with liver diseases causing HE
All stages Polyuria/polydipsia, vomiting, decreased endurance, inactivity, sometimes insufficient growth (e.
g., dogs with congenital shunts)General Periodic occurrence is very typical
growth factors; the expression of which requires the delivery of stimulating factors from the portal circulation. Thus, without these stimulating factors, the liver of animals with congenital shunts lack normal growth as the body grows. This explains why animals with congenital hepatic shunts most commonly develop symptoms when they are six months of age or older.
The clinical symptoms of HE vary and are due to metabolic derangements in the brain. If the underlying disease can be cured, even severe neurological signs disappear completely. Very characteristic is the episodic nature of HE, with fluctuations between grade one and the more advanced stages in the same patient (Table 7.2). Usually one or a few days of severe signs of HE alternate with more ore less normal periods of one or several weeks’ duration. Apart from the neurological symptoms of HE, non-neurological signs related to the underlying disease are often seen. Patients with HE rarely have seizures. Seizures without any of the other signs described in Table 7.2 are almost never due to HE.
Essentially, chronic HE is a dysfunction of several neurotransmitter systems. The most important ones involved are the glutamate, the dopamine /noradrenaline, and the gamma-aminobutyric acid and benzodiazepine (GABA / BZ) neurotransmitter systems. For the production and homeostasis of these transmitter systems, the brain utilizes precursors coming from the intestinal tract, which are normally modified by the liver. In patients with portosystemic shunting of blood, the lack of this modification gives these precursors unregulated access to the brain, which cannot adjust to the increased amount of unmodified neurotransmitters.
Glutamate is one of the most important excitatory neurotransmitters in the brain, and is directly influenced by the concentration of ammonia in the circulation (Figure 7.5). Ammonia is mainly produced in the intestinal tract by colonic bacterial degradation of nitrogenous compounds (i.e., proteins, amines, and urea) and also by the intermediary metabolism of glutamine in the mucosa of the entire intestinal tract.
The normal liver is extremely efficient in removing ammonia from the portal blood and in one passage of blood through the liver virtually all the ammonia is removed, so that the peripheral concentration is kept very low. Most of the ammonia is converted to urea by enzymes of the urea cycle of the hepatocytes located around the portal areas of the liver lobules; this cycle is exclusive to the liver. The urea is transported by the blood to the kidneys, which in turn excrete it into the urine. Another pathway of ammonia removal utilized by the cells of all tissues (in addition to the liver) is its incorporation in glutamate and glutamine. Glutamine contains two molecules of bound ammonia. Glutamine enters the circulation and becomes metabolized in the intestinal mucosa and the kidneys, where the ammonia is liberated. Intestinal ammonia reenters the cycle, but in the kidneys the ammonia produced in the tubular cells is excreted into the urine. However, in the case of alkalosis, ammonia can easily diffuse back into the renal vein, so that the kidneys then become a contributor to the plasma concentration of ammonia. In patients with portosystemic shunting of portal blood, the efficient removal by the liver largely fails and the plasma concentration of ammonia increases steadily.Hyperammonemia leads to toxic concentrations of ammonia in the nervous system because the physiological protection by the astrocytes becomes inadequate. The neurons are separated from the blood by a layer of astrocytes, and substances from the circulation have to pass these astrocytes before they can reach the neurons. Under physiological conditions, blood ammonia enters the astrocytes, but then gets scavenged by being incorporated into glutamine by a process that requires ATP and is catalyzed by glutamine synthetase. This enzyme has little functional reserve and cannot handle much more than physiological blood ammonia concentrations (Figure 7.5). The glutamine diffuses into the adjacent neurons where it is converted to glutamate by glutaminase. Glutamate in the neurons is then partly converted to GABA. The excitatory glutamate and inhibitory GABA form a finely tuned equilibrium determining the excitability of postsynaptic neurons. During times of hyperammonemia, the capacity of glutamine synthetase in the astrocytes becomes overloaded and free ammonia diffuses into the neurons. High neuronal ammonia concentrations inhibit glutaminase activity leading to the accumulation of glutamine and depletion of the neurotransmitter glutamate. This disturbed glutamate-glutamine-ammonia shuttle between the astrocytes and neurons is thought to be an important factor in the pathogenesis of HE.
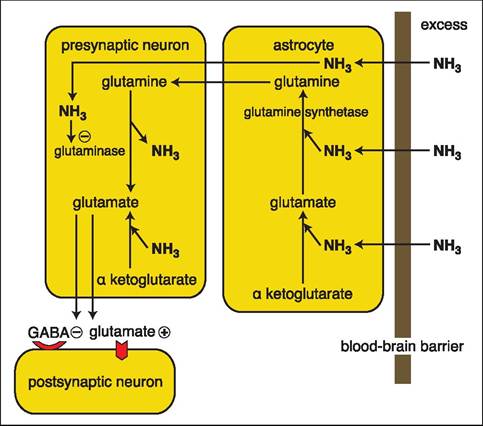
Figure 7.5:
Influence of hyperammonemia on glutaminergic neurotransmitters. Astrocytes are overwhelmed by the amount of ammonia diffusing into them and can no longer incorporate all the ammonia into glutamate and glutamine. The excess ammonia diffuses into the presynaptic neurons and leads to the formation of additional glutamate and the inhibition of glutaminase, which further increases the amount of glutaminergic neurotransmitters in the presynaptic neuron. In turn, this excess of glutaminergic neurotransmitters in the presynaptic neuron leads to overstimulation of the postsynaptic neuron. The minus sign indicates inhibition, while the plus sign indicates activation.
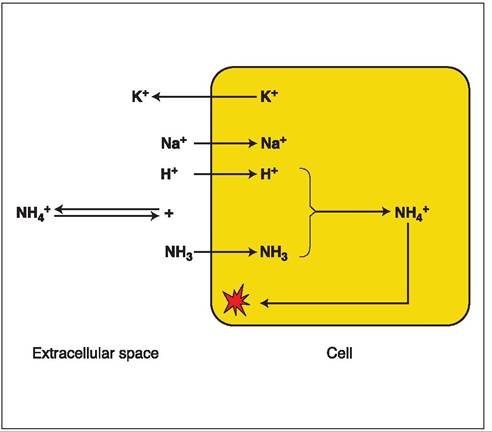
Figure 7.6:
Effect of hypokalemia on intracellular ammonia concentration. Hypokalemia leads to a movement of intracellular potassium into the extracellular space. In return, there is an influx of sodium and hydrogen ions. As a result, extracellular alkalosis ensues and leads to the generation of more ammonia and hydrogen from NH4+. Ammonia can easily diffuse into the cell. Within the cell, the excess hydrogen ions and ammonia form NH4+ once again. NH4+ can not diffuse back out of the cell and gets trapped.
Only the non-ionized form of ammonia, NH3, can pass through cell membranes, but NH4+ cannot. However, inside the neurons both forms are equally toxic. NH4+ and NH3, are both measured when blood ammonia is determined. In extra- and intracellular fluid, there is an equilibrium between NH4+ and NH3 + H+. This equilibrium shifts towards NH3 during alkalosis and towards NH4+ during times of a neutral blood pH or during acidosis. Thus, during alkalosis ammonia has easy access into the neurons, so that the same plasma concentration of ammonia may lead to a more severe encephalopathy than during times of a neutral blood pH or during acidosis. Therefore, alkalosis should be prevented or, if present, corrected. Alkalosis also results in the formation of alkaline urine, from which the non-ionized ammonia is readily reabsorbed, so that the kidneys may spare ammonia instead of excreting it. The most serious form of alkalosis is induced by hypokalemia (Figure 7.6). Low plasma potassium is replenished by an exchange of intracellular potassium against sodium and hydrogen ions. This exchange of hydrogen causes extracellular alkalosis and intracellular acidosis. As a result, ammonia can easily penetrate the cell membrane, but intracellularly it becomes ionized and can no longer leave the cell, so that the neurons further accumulate ammonia. Under these conditions, most of the ammonia pool is intracellular, and relatively moderate elevations in plasma ammonia concentrations may lead to rather severe neurological signs. Such conditions may very well occur in patients with chronic liver disease. The most frequent cause of this situation is portal hypertension, which in turn leads to ascites. The peritoneal fluid originates from the circulatory volume and many animals with ascites are slightly dehydrated, especially during the initial formation of ascites. This slight dehydration in turn activates the renin-angiotensin- aldosterone system, leading to renal sodium retention and potassium loss. In these patients, it is contraindicated to tap much ascitic fluid as the ascitic fluid will reform quickly. For the same reason, it is important to use only potassium-sparing diuretics, especially in anorectic patients that have insufficient potassium intake.
The measurement of blood ammonia concentration is currently the only practical way to diagnose HE.14,43-46 Mild to moderate hyperammonemia may sometimes be missed in a venous sample. When there is any doubt, an ammonia tolerance test may give more definitive results.47 This test is also
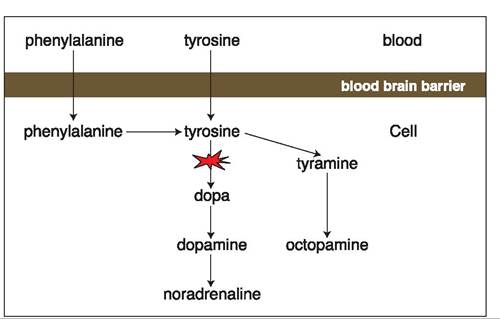
Figure 7.7:
Formation of false transmitters in patients with portosystemic shunts. In patients with portosystemic shunts, aromatic amino acids are not adequately cleared by the liver. As a consequence, more of these aromatic amino acids reach the neurons where they overwhelm the enzymatic capacity to form catecholamines and are instead metabolized to tyramine and octopamine. These “false” transmitters occupy the catecholamine receptors, but are non-functional and thus block mono- aminergic neurotransmission.
very reliable in diagnosing portosystemic collateral circulation in patients that do not yet have HE. The test is performed by giving a 5% ammonium chloride solution rectally (i.e., by advancing a red-rubber catheter approximately 10-20 cm into the rectum). Only in the case of portosystemic shunting does ammonia bypass the liver and peaks in the peripheral plasma at 20 and 40 min after administration. A distinct increase to above 100 and usually 150 μmol∕L is seen in patients with shunting of hepatic blood (reference range 150 μmol∕L). In approximately 40% of dogs with HE, ammonium biurate crystals can be identified during urinalysis.
In patients with HE, there is also an increased tone of the GABA/BZ receptor system. The underlying mechanism is poorly understood. However, the use of benzodiazepines and barbiturates that activate this receptor system is contraindicated in animals with suspected liver failure.
Additionally, in patients with portosystemic shunting, the aromatic amino acids tyrosine, tryptophane, and phenylalanine, which are absorbed from the intestinal tract, are not adequately cleared by the liver. If abnormally high concentrations of these amino acids reach the brain, an impairment of the catecholamine neurotransmitter system results. Tyrosine is the physiological precursor for the catecholamines (i.e., dopamine and noradrenaline), but the enzymatic capacity in neurons to utilize tyrosine is limited. In the case of an excess supply of tyrosine to the neurons, they produce alternative metabolites, such as tyramine and octopamine, which in turn occupy the catecholamine receptors, but are not functional (Figure 7.7). As a consequence, increased plasma tyrosine concentrations lead to a blockage of the catecholamine receptors in the central nervous system.
7.4.3.1 Management of hepatic encephalopathy
Mild cases of HE can be treated by feeding a low protein, high carbohydrate diet, which leads to a reduction of the ammonia and amino acid load. It is important to meet the energy requirements of the patient through dietary carbohydrates and fat as catabolism should be avoided in these cases. Cats require about twice as much protein as do dogs, which needs to be taken into account when choosing a diet. More severe cases of HE require treatment with the oral administration of lactulose (1-3 ml/kg/day divided over three doses), which is very efficient. Lactulose is not absorbed in the small intestine and is fermented to volatile free fatty acids by colonic bacteria. The resulting acidification shifts the equilibrium to non-absorbable ionized ammonia, increased colonic motility, and a less ammoniagenic colonic microflora. Also, a non-absorbable antibiotic, such as neomycin, may be useful in treating HE.
7.4.4 Coagulopathies
Because of the integral role of the liver in hemostasis, bleeding tendencies can be one of the presenting signs in cats and dogs with very severe hepatobiliary disease. However, the large functional reserve of the liver prevents clinical hemorrhagic symptoms in nearly all patients with even the most severe cases of liver disease. Most coagulation proteins and inhibitors, except for von Willebrand’s factor (vWF) and possibly factor VIII, are synthesized in the liver. An inability to synthesize vitamin K-dependent factors (i.e., factors II, VII, IX, and X) because of a failure of the bile acid-dependent fat absorption that can occur secondary to complete EBDO can cause coagulopathies. Subclinical coagulopathies may occur in diseases of the hepatic parenchyma.32,33
The most common cause of a subclinical coagulopathy in patients with hepatobiliary disease is disseminated intravascular coagulation (DIC). This is especially the case in patients with diffuse hepatocellular necrosis, as can occur in patients with hepatitis, lymphosarcoma, or metastatic tumors. Depending on the severity of the disease process (i.e., the amount of thromboplastin released from the necrotic tissue), the coagulopathy may be subclinical or may be clinically apparent. Findings on laboratory tests indicating DIC include a low concentration of
fibrinogen, thrombocytopenia, and the presence of fibrin degradation products. Therefore, it is essential to assess the coagulation status in patients with suspected hepatic disease, especially before collecting a liver biopsy. Fibrinogen concentrations below 100 mg/dl (1 g/L) are an absolute contraindication for a liver biopsy.
Although DIC can ultimately lead to blood loss, another mechanism for blood loss in dogs with severe hepatic disease is portal hypertension-induced vascular congestion and fragility. In such cases, blood loss may occur in the stomach or duodenum.
7.3.7 Polyuria and polydipsia
Increased thirst and volume of urination can be seen in patients with severe hepatocellular dysfunction. However, it should be noted that polyuria and polydipsia (PU/ PD) is a symptom of liver disease only in dogs, but not in cats. An increased sense of thirst may be a manifestation of HE. Also, excess secretion of ACTH from the intermediate pituitary lobe stimulated by abnormal neurotransmitters can lead to excess cortisol secretion from the adrenal glands and an altered threshold for antidiuretic hormone release in dogs with HE.44 However, PU/PD also frequently occurs in dogs with liver diseases that are not associated with portosystemic shunting. Although several mechanisms have been proposed, the pathophysiology is still not entirely clear.
7.4