Equine Grass Sickness
Robert J. MacKay
Grass sickness is a debilitating, often fatal, dysautonomia of equids associated with full-time access to pasture.1 Because lesions are also found in the somatic lower motor neurons, it has been suggested that it is more accurate to describe grass sickness as a polyneuropathy rather than a dysautonomia.2 A comparable fatal polyneuropathy has also long been recognized in the Patagonia region of South America, where it is known as mal seco? Equine grass sickness (EGS) is endemic in Scotland, England, and Wales and has also been reported from many countries in northern Europe, Cyprus, the Falkland islands, and Colombia with isolated cases in Australia, Ethiopia, and the United States.1,4-6
Epidemiologic studies conducted since the early 1970s have identified high horse density, full-time grazing, a recent change in feed type or premises, previous occurrence of the disease on the premises, pasture disturbance, mechanical removal of horse manure from pastures, sandy or loamy soils, recent deworming with ivermectin, recent cool dry weather and irregular ground frosts, and good bodily condition as risk factors.1,2,7-14 Concentrations of various herbage and soil minerals are associated with horses' risk of developing EGS.2 Cases occur throughout the year with peak numbers in the spring and a smaller peak in the autumn.
The risk of disease is highest in horses aged 2 to 7 years (peak, 5 to 6 years), although cases have been reported in horses from 2 months to 27 years of age. Horses of all breeds, ponies, donkeys, Przewalski's horses, and zebras are affected. Males and females are equally represented in surveillance data. Protective factors included contact with previous cases, daily feeding of hay or haylage, chalky soil, higher average maximal temperature, manual removal of feces from pastures, pasture grass mowing, and grazing together with ruminants.■ Cause and Pathogenesis Theories as to the cause of EGS have included mineral or vitamin deficiencies or exposure to toxic plants, secondary metabolites and trace minerals in pasture grass, insects, viruses, or bacterial or fungal toxins.15,16 Although the cause of EGS is still unknown, considerable evidence has been adduced in support of roles either for C. botulinum toxins or mycotoxins.
The hypothesis that mycotoxicosis is the cause of EGS is attractive because it is consistent with the observed influences of pasture grazing and climate on disease risk.2 Since the 1990s, a hypothesis has been developed that EGS is a toxicoinfectious form of botulism involving a type C strain commonly carried by birds.17 Strong support for this hypothesis has been provided by (1) the demonstrated association between EGS and the presence in ileal contents of C. botulinum type C and type C1 neurotoxin (BoNT type C)17; (2) the observation that horses with EGS have significantly lower serum antibodies to C. botulinum and BoNT type C than do pasturemate controls18,19; and (3) the presence of rising titers of specific IgG antitoxin in cases of chronic EGS. C. botulinum type C is unique among clostridial organisms in that its major toxin, BoNT type C, possesses potent and nonspecific neurotoxicity in vitro, probably via action on cellular syntaxin.17 It has been hypothesized that toxin production and absorption occur in the ileum as a result of either overgrowth from normal intestinal flora or spore germination in association with nutritional or environmental triggers involving factors such as parasite status and changes in feed, pasture, and weather conditions.1,4 Set against this evidence are the many clinical, epidemiologic, and biological differences between EGS and neuroparalytic botulism of horses.2 A survey of veterinarians in Great Britain confirmed the feasibility of conducting a controlled field trial of the efficacy of C.
botulinum type C vaccination in preventing naturally occurring EGS.20 A candidate vaccine was selected and shown to induce significant antitoxin responses and a longitudinal vaccine study has been ongoing since 2014.21■ Clinical Signs The major clinical signs of grass sickness principally reflect dysfunction of the enteric nervous system.1,22 They include dysphagia, gastric and small intestinal dilation, nasogastric reflux, colonic impaction, and colic. Clinical presentations are classified as acute (course of 7 days).23 Two thirds of reported cases have been acute or subacute and one third was classified as chronic.1 Acutely affected horses may be found dead or in abdominal crisis with signs of colic, tachycardia, generalized sweating, nasogastric reflux (including spontaneous nasal regurgitation of stomach contents), absence of intestinal borborygmi, and abdominal distention. Rectal palpation and transabdominal ultrasonography reveal distended loops of small intestine and gas-filled large bowel. Affected horses are unable to swallow food or water, and drool from the mouth may be in the form of thick, ropy saliva. Fine muscle fasciculations similar to those that occur with EMND may affect the triceps and quadriceps.
Horses with subacute EGS survive long enough to develop colonic impactions and secondary large intestinal tympany. Chronic EGS may develop insidiously, and only a minority of these cases show mild, intermittent colic. Patchy sweating or piloerection, or both, are evident on the neck, flanks, and behind the shoulders. Typically, the appetite is reduced, and affected animals have varying degrees of difficulty in chewing and swallowing; however, salivation, accumulation of fluid in the stomach, and impaction are not usual features. Bilateral ptosis with ventral deviation of the eyelashes are often secondary to loss of sympathetic eyelid tone. Because of suppression of normal nasal secretions, some horses with mild chronic EGS have a characteristic dry crusty nasal exudate (rhinitis sicca).
Fecal production is reduced, and feces are unusually firm and small. Weight loss is rapid and severe.Acute and subacute cases are invariably fatal. Death occurs from gastric rupture, circulatory compromise, or cardiopulmonary failure; most commonly, such animals are euthanized. With appropriate nursing case, many horses with chronic (mild) EGS survive and even completely recover.2 EGS is unlikely to recur in recovered horses.13
■ Diagnosis Definitive diagnosis requires demonstration of compatible neuropathologic features within the autonomic or enteric nervous systems.1 The antemortem sample of choice is an ileal biopsy, which is processed for hematoxylin and eosin staining with or without synaptophysin immunostaining of myenteric and submucosal enteric plexuses.2 Lingual and rectal biopsies have been undertaken for diagnostic accuracy, and preliminary results have been promising but require prospective validation.2
No confirmatory blood test for EGS is available. Nonspecific abnormalities are consistent with severe dehydration, systemic inflammatory response, hypovolemia, stress, loss of GI secretions, and anorexia.2,24 Peritoneal fluid protein concentrations may be high, although nucleated and red blood cell counts are usually normal. Altered serum concentrations of numerous acute-phase proteins (including α2 macroglobulin, serum amyloid A, ceruloplasmin, haptoglobin, and orosomucoid) have been recorded in horses with EGS but are nonspecific indicators of inflammation.25
In EGS cases with ptosis, topical application of 0.5 mL 0.5% phenylephrine eyedrops results in normalization of the eyelash angle within 30 minutes, and this result is supportive of the diagnosis.26 Barium swallow contrast radiography, esophageal endoscopy, abdominal ultrasonography, and rectal palpation are used to identify changes consistent with dysfunction of components of the enteric nervous system.2
■ Treatment and Prevention All cases of acute and subacute EGS are considered incurable and therefore necessitate euthanasia on humane grounds as soon as possible after diagnosis.
For selected chronic cases, treatment involves excellent nursing care and provision of palatable, easily swallowed food and high energy concentrates. Clinical recovery occurs in approximately 70% of preselected treated cases.1Current efforts at prevention involve minimization of known risk factors and introduction of suspected protective practices. Thus in endemic areas, horse owners are advised to avoid exposure to pastures where previous cases have occurred, minimize disturbance of pasture or soil, prevent overgrazing of pasture, and avoid the “overuse” of ivermectin dewormers. On the other hand, owners are encouraged to allow horses to graze with ruminants, mow pastures regularly, manually remove feces from pastures, and supplement pasture feeding with hay or haylage. These practices are particularly important during seasonal and climatic risk periods, as well as for young horses in good bodily condition, especially those recently imported.
■ Necropsy Findings Gross necropsy findings in horses with EGS include esophageal ulceration, fluid distention of the stomach and small intestines, impaction of the large colon or cecum (or both), and the presence of inspissated mucus within the small colon and rectum. Horses with subacute and chronic cases do not have gastric and small intestinal distention but do have variable degrees of large intestinal impaction and often rhinitis sicca. Histologic lesions of neuronal degeneration are most evident in the autonomic ganglia, enteric nerve plexuses, brain, and spinal cord.23,27 There is chromatolysis, nuclear eccentricity, nuclear degenerative changes, cell death and neuronophagia, cytoplasmic vacuolation, and spheroid formation.
Peripheral Nerve Disorders can establish competent junctions with adjacent denervated muscle units. Reinnervation of muscle units by this process occurs within days to weeks. However, if the motor axons to a muscle are severed, reinnervation occurs by growth of collateral sprouts from the proximal stump.
Axonal sprouts grow at a rate of 1 mm per day (≈1 inch per month). Because of progressive fibrous replacement of denervated muscle and retrograde degeneration of the proximal parts of affected motor neurons, reinnervation may not be possible if more than 12 months have elapsed since the original injury. However, reinnervation surgery has been successful when performed on human patients 20 years or more after denervation.2Large-diameter myelinated fibers are most sensitive to compression forces. The largest axons (sensory axons), including afferent fibers conveying touch-pressure, vibration, and proprioception, are usually damaged first (which results in ataxia or inappropriate limb placement); second, the large myelinated motor axons are compromised; third, the smaller axons, which carry cutaneous and deep mechanoreceptor sensory information, are affected; last to be lost are the smallest-diameter fibers, which transmit pain sensation.
The following sections are discussions of the peripheral nerves most often damaged in large animals.
Suprascapular Nerve
Mechanical damage to the suprascapular nerve results in paralysis of the infraspinatus and supraspinatus muscles.3 Such injuries occur most commonly when the animal collides at speed with gateposts, trees, or other objects.4 Acute injury or local anesthesia of the suprascapular nerve is characterized by outward bowing or “popping” of the scapulohumeral joint, or both, as weight is placed on the limb. Neurogenic atrophy develops after 2 to 4 weeks, and the scapular spine becomes prominent (Fig. 35.24). The common name for this condition is “sweeney.”
Brachial Plexus
Damage to the brachial plexus may result in any combination of dysfunction of the radial, pectoral, axillary, musculocutaneous, median, and ulnar nerves, although radial nerve signs usually dominate.5,6
Lesions of the brachial plexus are caused by trauma to the shoulder with or without scapular or rib fracture, deep penetrating axillary wounds, neoplasia or bacterial cellulitis, or forced extraction of a fetus to relieve dystocia.7 Because of their
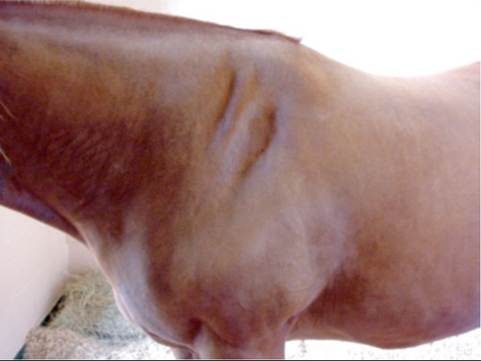
FIG. 35.24 Sweeney (neurogenic atrophy) of the left supraspinatus and infraspinatus muscles in a Quarter Horse gelding. The lesion was caused by trauma related to running into a fencepost 2 months previously.
tendency to move at high speeds, horses are most susceptible to brachial plexus injuries. The condition may occur in small ruminants after automobile accidents, carnivore attacks, or blows from larger animals.
Avulsion of the brachial plexus results in complete flaccidity, lack of reflexes, and desensitization of the affected thoracic limb. Disruption of the origins of the cervical sympathetic trunk in the caudal aspect of the plexus results in ipsilateral Horner syndrome. Lesser injuries may result in abduction of the elbow (pectoral nerve), dropped shoulder (subscapular), hyperextension of the elbow (musculocutaneous), or signs of radial paralysis (next section).8
Radial Nerve
The radial nerve provides motor input to a flexor of the shoulder and extensors of the elbow, carpal, and digital joints. The nerve courses over the lateral aspect of the elbow joint and is vulnerable to traumatic insult at that point. Radial nerve paralysis arises from injury to the T1 nerve root or nerve from vertebral or rib fracture, as part of a brachial plexus injury, as a result of humeral fracture, from direct trauma to the nerve over the elbow, as part of an ischemic myoneuropathy during prolonged anesthesia, or during restraint in lateral recumbency with inadequate padding of the forelimb.7,9-11
With radial paralysis, the limb position varies, depending on the location of the lesion in the radial nerve. Lesions at or near the elbow joint are characterized by dropped elbow, inability to protract the limb, scuffing of the toe, and flexion of all distal limb joints (Fig. 35.25). The foot is knuckled over at rest, and the animal is unable to bear weight on the leg. Lesions distal to the elbow cause knuckling of the carpus, fetlock, and pastern joints, but the animal can support weight on the affected limb if the metacarpus and distal limb are held in extension. The triceps reflex is depressed or absent. After weeks of radial denervation, the extensor muscles of the thoracic limb exhibit atrophy. Detectable sensory deficits resulting from radial nerve paralysis tend to be vague and probably vary from patient to patient.
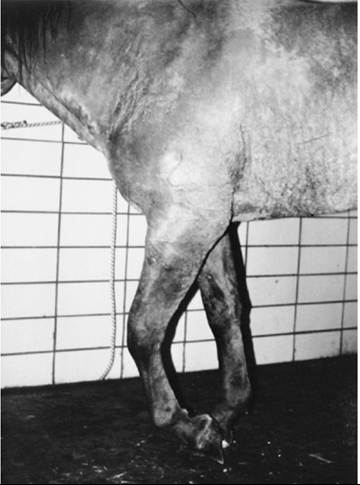
FIG. 35.25 High radial nerve paralysis in a horse showing knuckling of the carpus and digit and dropped elbow.
KANGAROO GAIT IN SHEEP. A locomotor disorder predominantly involving the thoracic limbs of lactating ewes has been reported in New Zealand, Australia, Scotland, and northern England.12,13 Knuckling and paresis of the thoracic limbs are suggestive of radial paresis, and a high-stepping forelimb gait is also present. In advanced cases, the pelvic limbs are also involved. Limited necropsy data suggest both CNS and peripheral nerve involvement.14,15 Affected ewes are usually of large breeds and are nursing twins. Most recover after weaning their lambs. Affected sheep are usually grazing on mixed forage, often with some concentrate supplementation. A clinically identical condition has been reported in Israel and in Australia among sheep grazing on the legume fenugreek.16 A causative toxin or nutritional deficiency is suspected for both kangaroo gait and fenugreek staggers, but no toxin has yet been identified.
Femoral Nerve
The femoral nerve is distributed to the quadriceps femoris muscles and the skin of the rear limb over the medial thigh. Traumatic overextension of the hip and stifle joints from a fall or other injury or forced posterior delivery of a fetus can damage one or both femoral nerves.17 The nerve courses too deeply to be directly affected by an external blow to the pelvic limb; however, it can be damaged by ilial, femoral, or vertebral fractures. Ischemic injury caused by prolonged stretch or increased tissue pressure during anesthesia in dorsal recumbency may cause bilateral femoral neurapraxia.18 A similar presentation, presumably caused by compression of lumbar spinal branches by the foal in the birth canal, occurs in mares after severe prolonged dystocia.
The clinical signs of femoral nerve paralysis are related to an inability to extend and fix the stifle.3 The reciprocal apparatus is unable to fix the stifle and hock, which results in collapse of the limb during weight bearing and constant flexion of all distal digital joints. The hoof usually remains flat on the ground. Chronic femoral denervation causes atrophy of the quadriceps femoris muscles. The patellar reflex is absent or depressed, and the patella is often displaced laterally. Hypoesthesia or anesthesia of the medial part of the rear limb extends from the proximal thigh to the medial malleolus of the tibia.
Sciatic Nerve
The sciatic nerve is the largest nerve in the body and innervates the extensor muscles of the hip, flexor muscles of the stifle, and most of the muscles of the distal limb. Sciatic nerve paralysis occurs most often in postpartum cows after forced fetal extrac- tion.19 Injury to the sixth lumbar nerve proximal to the lumbosacral plexus is the usual cause of so-called calving or obturator paralysis syndrome. Injection of irritating drugs into the space between the greater trochanter and the ischial tuberosity may cause a sciatic neuritis.20 This occurs most frequently in young animals, but rare cases occur in adult cattle and small ruminants. Other causes of sciatic nerve damage are pelvic fractures, sacroiliac or coxofemoral dislocations, tumors, or abscesses located along the course of the nerve. Sciatic neuropathy is a rare complication of sacroiliac joint injections in horses.
The sciatic nerve innervates most of the musculature of the rear limb, and so the motor deficits associated with denervation are profound. At rest, the limb hangs behind the animal. The stifle is dropped and extended, and the hoof rests on its dorsum (Fig. 35.26). If the limb is positioned properly, the animal usually can bear some weight on it because of the normal function of the quadriceps muscles and the action of the reciprocal apparatus. Chronic sciatic denervation results in neurogenic atrophy of the caudal thigh muscles and all of the muscles distal to the stifle.
The sciatic nerve divides into the tibial and the peroneal nerves in the distal limb. Therefore except for the medial
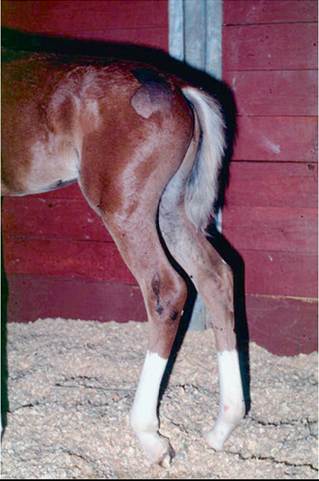
FIG. 35.26 Characteristic posture of a foal with partial sciatic nerve paralysis. Note the flexion of the hocks, fetlocks, and stifle. The signs were the result of an intramuscular injection in the gluteal area.
part of the thigh and gaskin, sciatic denervation also results in hypoesthesia and anesthesia of the entire limb distal to the stifle.
Peroneal Nerve
The peroneal branch of the sciatic nerve is distributed to the flexor muscles of the hock joint and the extensor muscles of the digit. The nerve becomes superficial and is exposed to damage as it crosses over the lateral condyle of the fibula immediately distal to the stifle. Peroneal paralysis of this type occurs in all species of large animals and is common in postpartum dairy cattle that have been recumbent as a result of hypocalcemia or other causes and in horses because of postanesthetic ischemic myoneuropathy. The hock joint is hyperextended, the fetlock and pastern are flexed, and the dorsum of the hoof usually rests on the ground3,21,22 (Fig. 35.27). Affected animals can bear weight when the limb is manually placed in the proper position. There is desensitization of the skin over the craniolateral aspect of the limb extending from the stifle to the hoof.
Tibial Nerve
The tibial nerve supplies the extensor muscles of the hock joint (i.e., the gastrocnemius) and the digital flexors. Tibial paralysis is most often observed in periparturient cattle or neonates that have been given an injection of an irritant drug in the caudal leg at the level of the stifle. Tibial paralysis also occurs in sheep and goats and is a common sequela to dog-bite injuries. The resting limb is held more flexed than is normal, and, although the foot contacts the ground in normal position, the metacarpophalangeal joint often partially collapses into a flexed position (i.e., “knuckles”). The limb moves in a stringhaltlike manner, with exaggerated flexion of the tarsus and stifle during protraction, followed by sudden extension to the weight-bearing phase of the stride. At rest the pelvis is asymmetric, with the affected side held lower than normal.3 Chronic loss of tibial nerve function results in atrophy of the gastrocnemius and digital flexor muscles. The skin of the caudomedial aspect of the leg demonstrates anesthesia.

FIG. 35.27 Calf with peroneal nerve paralysis. Note the flexed fetlock and digit and the hyperextended carpus. This condition was caused by injection of antibiotics into the peroneal nerve on the caudolateral aspect of the leg.
Obturator Nerve
The obturator nerve supplies the adductor muscles of the pelvic limb. The nerve is well protected in horses and small ruminants, and so injury is rare in these species. In contrast, because the cow has a relatively shallow acetabulum and poorly developed round ligament, the obturator nerve and the lumbar branches that supply it are vulnerable to injury during forced extraction of a calf to relieve dystocia. Reports of obturator nerve paralysis of cattle accompanied by knuckling and inability to support weight on the rear limbs probably represent a combination of sciatic and obturator nerve dysfunction. Coxofemoral dislocation is a common complication of obturator paralysis in cattle housed in stalls with slippery flooring.
Obturator nerve paralysis is most common in cattle and is almost exclusively a result of dystocia. The nerve injury is located in the pelvis at the level of the obturator foramen. Of all dystocias in dairy cattle, 9.2% result in paraplegia.23 The obturator nerve innervates the adductor, pectineus, and gracilis muscles. Effects are minimal if the cow is placed on a surface that has good traction. Clinical signs of an obturator nerve injury include a hopping pelvic limb gait when the animal attempts to run and severe pelvic limb abduction or splay-leggedness when the animal stands on a slippery surface (Fig. 35.28). In severe cases, the cow may be sternally recumbent with the pelvic limbs extending laterally to each side. Experimental studies have indicated that the so-called calving paralysis syndrome is actually a result of a combination of injuries to the obturator nerve and to the sixth lumbar spinal nerve (L6), a major contributor to the sciatic and obturator nerves. Experimental sectioning of the obturator nerve alone does not produce paralysis, if the animal has secure footing.3,19,24 Obturator nerve paralysis does not result in a cutaneous sensory deficit.
Compression damage to one or more peripheral nerves during parturition or milk fever is a frequent factor in the so-called downer cow syndrome (discussed later). The condition occurs in 4 to 28% of all cases of milk fever and, before the advent of flotation tanks for treatment, was associated with a mortality rate of 20 to 67%.25,26
Peripheral Facial Nerve Paralysis
Facial paralysis is common in horses but uncommon in other large animals. Depending on the site of damage, some or all

FIG. 35.28 Cow with obturator nerve paralysis caused by relief of a difficult dystocia. Note the basewide stance and yet the apparent ease with which the cow is able to stand on the deep bedding. Note also that rope hobbles are being applied to prevent further abduction and trauma.
of the facial muscles can be affected. Complete unilateral facial paralysis in horses is evident as deviation of the nose toward the normal side, reduced flaring of the ipsilateral nostril during inspiration, and ipsilateral drooping of the lip, eyelid, and ear. Reflexes involving cranial nerve VII, such as the lip, eyelid, and menace and ear reflexes, are reduced or absent. Inability to close the eyelid causes exposure keratitis, which may be particularly severe when there is damage to the secretomotor fibers of the facial nerve at or proximal to the geniculate ganglion (keratitis sicca). The tongue protrudes out of the paralyzed side of the mouth, which suggests, often erroneously, a problem with lingual function. Because of paralysis of the buccinator cheek muscle, boluses of feed accumulate between the cheek and teeth, from where they may be dropped from the mouth. With chronic facial denervation in horses, neurogenic atrophy of the parotidoauricularis muscle becomes apparent as a groove behind the vertical ramus of the mandible. In cattle, because of the rigid structure of the planum nasale, muzzle deviation is not observed. In small ruminants, subtle muzzle deviation can usually be appreciated.
Injury to the facial nerve proximal to the vertical ramus of the mandible causes full facial paresis. Proximal facial nerve trauma in horses is usually caused by fractures of the vertical ramus of the mandible, stylohyoid bone, or petrous temporal bone (after a blow to the head or as a complication of THO). Damage within the facial canal is often associated with concurrent signs of vestibular nerve damage such as head tilt, nystagmus, and circling. Tear production may also be reduced or absent. Hemorrhage into the middle/inner ear, otitis media/ internal guttural pouch mycosis, and parotid lymph node abscessation each can involve the adjacent facial nerve, causing ipsilateral facial paralysis. Distal facial nerve damage is usually due to direct injury from a blow or lateral recumbency or strangulation by a tight-fitting halter. The nerve or its branches are often damaged as they cross the mandible, the zygomatic arch, or both. Facial paresis after recumbency during general anesthesia is common and usually involves only the nose, lips, or both. Similar conditions occur in goats and are caused by excessive pull on neck chains while the animal is being led onto the milk stand or tied with the neck chain. In cattle, facial nerve paresis is usually the result of space-occupying masses at the caudal aspect of the mandibular ramus.
Most animals with facial nerve paralysis caused by trauma have neurapractic (type 1) nerve injury and recover within 2 weeks; those with type 2 (axonotmetic) injuries regrow axons from the site of injury at the rate of approximately 1 inch per month, and so full resolution make take several months. The paralysis is occasionally permanent. In chronic cases, food should be removed from the cheek pouches twice daily, and the tongue and fauces should be routinely examined for ulcers. Oral ulcers, if present, should be treated by flushing of the mouth.
■ Treatment The objectives of medical treatment of peripheral nerve injury are (1) to reduce swelling and inflammation at the site of injury, (2) to support and stabilize the denervated area, and (3) to maintain of the general health of the animal.
In the case of lacerating trauma, the general principles of wound management apply. The wound should be cleaned, irrigated, debrided, and dressed, and ventral drainage should be established. If severed nerve ends are visible, they can be tagged for later identification and surgical repair. Medical therapy for inflammation and swelling should be initiated, especially if there is obvious tissue swelling. This may include any combination of a cyclooxygenase inhibitor (e.g., flunixin meglumine, 1.1 mg/kg IV, IM, or PO once or twice daily); DMSO (1 g/kg as a 10% solution IV or by nasogastric tube once or twice daily); and a glucocorticoid (e.g., dexamethasone, 0.05 mg/kg IV or IM once daily). An additional antiinflammatory effect can be provided by cold water hydrotherapy or local application of skin-permeant drugs such as DMSO or diclofenac. In the presence of an open wound or other risk for bacterial infection, broad-spectrum antimicrobial therapy should be initiated. This can be directed by culture results, if available.
Fractured or dislocated bones that have the potential to further injure nerves must be reduced and stabilized. Stall rest helps prevent further injury. In the case of paralyzed limbs, a protective wrap may be necessary to prevent damage caused by dragging or improperly placing the limb. A support wrap, splint, or light cast should be maintained on a leg affected with radial paralysis so as to prevent limb contracture. A heavy support wrap, possibly with additional fetlock support, should also be placed on the opposite limb to protect against breakdown caused by prolonged increased weight bearing. If the horse has difficulty alternating between recumbency and standing, it should be supported as needed in an abdominal sling (horses and small ruminants) or flotation tank (cattle) until it is able to rise unassisted. Because of the buoyancy provided by water, affected animals (usually horses) may be able to exercise affected limbs in swimming pools. Skin over bony prominences should be padded to prevent decubital injury; such injuries must be treated aggressively if they occur.
If possible, passive range-of-motion exercises should be performed on paralyzed limbs in an attempt to preserve joint flexibility and prevent contracture. Faradic or galvanic electrical muscle stimulation, acupuncture, and hyperbaric oxygen therapy are other procedures advocated for treatment of peripheral nerve injury. Calcium gluconate can be given empirically to downer cows (500 mL SC daily), as may potassium chloride (100 g in 20 L of water given daily by stomach tube). Abduction of the rear limbs may be prevented in cattle by application of a hobble around the metatarsus. The distance between the legs should be approximately 35.6 to 50.8 cm (14 to 20 inches) when the legs are tied together and held abducted. It is important to apply the hobbles with a nontightening bowline knot to prevent strangulation of the foot.
Surgical procedures for repair of peripheral nerves are beyond the scope of this text but are described elsewhere. In external neurolysis, the nerve is dissected from a bed of scar tissue.1 This is indicated if there is obvious compression from fibrous tissue, and it is performed routinely in horses with suprascapular neuropathy.