Genotyping Schemes and Population Structure of Major Zoophilic Dermatophytes
Genotyping is often employed to confirm or rule out outbreaks, gain insight into the dynamics of disease transmission, recognize virulent strains and regional and global changes in genotype pattern, determine the source and routes of infections, trace cross-transmission of healthcare-associated pathogens, and evaluate the effectiveness of control measures (Ranjbar et al.
2014). Other common issues in dermatophytes concern differentiation of relapse or reinfection, determining if the infection is caused by one or more strains, if the host can harbor different genotypes varying in their degree of virulence or potential for transmission to other hosts including human, if particular genotypes differ in clinical manifestation, etc. The success in typing of dermatophytes according to phenotype criteria has been limited. Indeed, the same strain may present with different phenotypic characteristics depending on different factors, firstly the conditions of culture (Dhieb et al. 2014).Fig. 3.3 (continued) branches were obtained from 1000 bootstrap replicates. Only branches with bootstrap support ≥70% are shown; branches with support ≥95% are double thick; ex-type strains are designated by a superscript T. The tree is rooted with Ctenomyces serratus On the other hand, strains with similar colonies may belong to different genetic types.
Because many pathogenic dermatophyte species show nearly clonal population structure and phenotype commonly do not correlate with genotype, DNA-based approaches are supposed to be tools of choice for genotyping. Although DNA sequencing allows species identification of dermatophytes, it lacks sufficient discriminatory power to study population structure of most clinically relevant species. Consequently, no multilocus sequence typing schemes (MLST) have been evaluated and developed for genotyping of dermatophytes, although MLST has found wide application in many other fungal pathogens (Meyer et al.
2009; Debourgogne et al. 2012; Bernhardt et al. 2013; Maitte et al. 2013). It is worth mentioning that certain level of intraspecies polymorphism was detected by using DNA sequencing of some genetic loci in sexual species, especially T. mentagrophytes and T. benhamiae. Four ITS rDNA sequence genotypes were revealed among 86 isolates of T. mentagrophytes sensu lato (Heidemann et al. 2010); genotyping was useful for discrimination between strains of zoophilic origin (T. mentagrophytes s. str.) and closely related anthropophilic T. interdigitale and correlated with clinical manifestation and phenotype of strains. Additional ITS rDNA genotypes in T. mentagrophytes s. str. were revealed by Pchelin et al. (2016) who did not confirm correlation between genotype, origin of strains, and phenotype observed by Heidemann et al. (2010). Combination of ITS rDNA and GPD gene revealed five genotypes in global population of T. benhamiae (Cmokova 2015) which corresponded with phenotype and in part with geographic origin. Apart from ITS and GPD genes, also TOP II (Kawasaki et al. 2011) and TEF-1α (Mirhendi et al. 2015) have potential for genotyping of T. mentagrophytes and T. benhamiae but are not available or lack discriminatory power in other pathogenic species. In conclusion, the set of four last mentioned genes has significant potential for typing of T. mentagrophytes and T. benhamiae, while population of M. canis and T. verrucosum in domestic animals and humans shows high level of clonality and resists to genotyping by currently available genetic loci. Limited variability was found in ITS region of M. canis (Kaneko et al. 2011); unfortunately significant part of the sequence variability in GenBank is caused by sequencing errors as demonstrated by at least four different ITS sequences deposited for the ex-type strain of M. canis.Microsatellite markers are currently the most powerful and effective tool available for subtyping of dermatophytes. The typing schemes have been developed for only limited number of species, i.e., T.
rubrum, Nannizzia (=Microsporum) persicolor, M. canis, and T. benhamiae. These methods will be given the most attention in the following paragraphs.Genotyping attempts were commonly unsuccessful in M. canis by using various approaches including RAPD, analysis of NTS region, or PCR-RFLP targeting ITS region (Yu et al. 2004; Leibner-Ciszak et al. 2010; Dobrowolska et al. 2011; Dhieb et al. 2014). In contrast, Spesso et al. (2013) was able to reveal intraspecies variability by using RAPD method among Argentine strains without significant correlation with clinical manifestation or geographic origin. Another method, inter-single-sequence-repeat-PCR (ISSR-PCR), revealed 21 genotypes among a total of 24 strains analyzed (Cano et al. 2005), which may indicate a good discriminatory power of the method employed. However, the stability of the markers was not assessed, and the reproducibility of the method was low. Two microsatellite loci were originally developed by Sharma et al. (2007), while more recently an extended panel of eight loci has been standardized (Pasquetti et al. 2013). By using two microsatellite loci, Sharma et al. (2007) studied genetic variation and dispersal among 101 global M. canis strains, distinguished three subpopulations, and found no correlation between genotype, clinical manifestation, and geographic origin. It was suggested that imbalance in the prevalence of particular genotypes among humans and animals was due to the emergence of a virulent genotype with a high potential to infect human host. Extended panel of eight loci proved to have a high discriminating power and revealed the extensive genetic diversity in global population of M. canis (Peano et al. 2015). Some multilocus genotypes (ML-GTs) were found with higher frequency, which leads to hypothesize the existence of clonal lines of “major success” due to a stronger parasitic aptitude. Correlation was not found between severity of clinical forms and a particular genotype. Likewise, there was no particular association between specific ML-GTs and zoonotic potential.
Some ML-GTs were related to specific geographical contexts. Although it is unlikely that the loci employed for strain typing are connected with phenotypical features of interest (such as virulence, drug resistance etc.), microsatellite analysis has the potential to track these features indirectly, principally due to the clonal mode of reproduction typical of most dermatophytes (genomes are transmitted to the next generation in unaltered condition and thus associated genes - such as virulence genes and microsatellite markers are linked, which may allow tracing the feature of interest within populations of the fungus). Hence, for example, microsatellites were used to demonstrate that isolates of M. canis causing pseudomycetoma in cats are genetically related, if not identical, to isolates responsible for superficial ringworm lesions. These results strongly suggest that pseudomycetoma in cats is due to host factors and cannot be attributed to the specific ability of particular genotypes (Pasquetti et al. 2012). The data from two previously published studies (Sharma et al. 2007; da Costa et al. 2013) were reanalyzed here. The results (Fig. 3.4) confirmed that there is no clear association between particular haplotypes, geographical origin, and host.Polymorphisms in population of T. benhamiae (American-European race) were investigated by using RFLP analysis of NTS region which produced 11 different patterns in 46 isolates; this method successfully confirmed laboratory-acquired infection as well as familial outbreaks transmitted from pets (Mochizuki et al. 2002; Takeda et al. 2012). The data indicated that T. benhamiae had been brought into Japan with imported animals on several occasions and spreads in Japan by transportation of animals by breeders or pet shops (Takeda et al. 2012; Hiruma et al. 2015). The global population structure of T. benhamiae (n = 326 isolates) was recently investigated by using sequences of two genetic loci (ITS rDNA and GPD) along with ten microsatellites markers (Cmokova 2015).
The combined sequence analysis revealed the presence of five genotypes, while 32 unique genotypes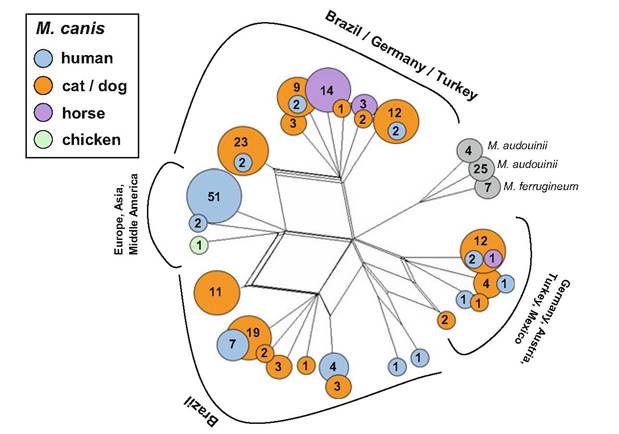
Fig. 3.4 Population structure of Microsporum canis complex revealed by analysis of two microsatellite loci—reanalysis of data previously published by Sharma et al. (2007) and da Costa et al. (2013). The dataset included 203 strains of M. canis, 29 M. audouinii, and 7 M. ferrugineum. NeighborNet phylogenetic network built using Jaccard index-based distance matrix with SplitsTree 4.13 (Huson and Bryant 2006). Colored circles correspond to different hosts; numbers in circles indicate number of isolates representing particular haplotype. No clear association between particular haplotypes, geographical origin, and host is evident. However, several haplotypes of “major success” were found in some hosts (haplotype represented by 51 human isolates; haplotype represented by 14 isolates from horses)
arranged into four subpopulations were discovered by microsatellite analysis (Fig. 3.5). The first subpopulation (S1) was most abundant in Europe and associated with guinea pigs; it was characterized by low variability in microsatellite data, yellow colonies with yellow reverse, and exclusively MAT1-1-1 idiomorph. This clonal subpopulation is currently responsible for the outbreak of infections in the Central Europe. The second subpopulation (S2) comprised strains from North America mostly associated with dogs and typical by highly variable microsatellite data, both mating-type genes were present among strains; the colonies were mostly white, granular, and frequently with red reverse. It is probable that virulent European subpopulation has its origin in closely related subpopulation S2 in North America where the center of genetic variability of T. benhamiae is located and where the pathogen probably occurs on wild animals. The third subpopulation (S3) comprised strains from Europe mostly associated with guinea pigs and characterized by the presence of both mating types (predominantly MAT1-2).
The fourth subpopulation (S4) comprised majority of strains from Japan, and some European strains that were mostly associated with rabbits; all strains had only MAT1-1 idiomorph. Some of these subpopulations could represent separate taxonomic entities, but more research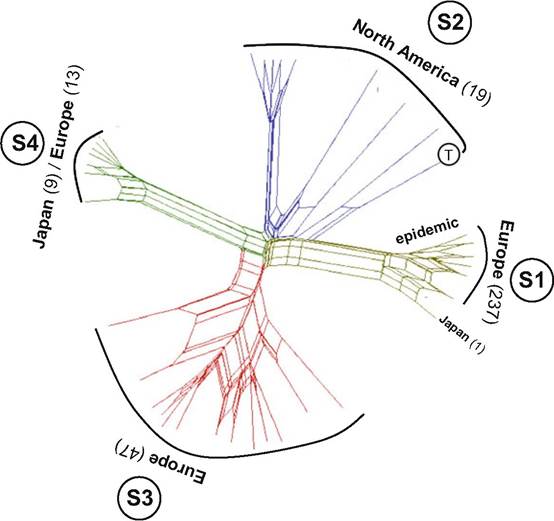
Fig. 3.5 Population structure of Trichophyton benhamiae (n = 326) revealed by analysis of ten microsatellite loci (Cmokova et al. 2017). NeighborNet phylogenetic network built using Jaccard index-based distance matrix with SplitsTree 4.13 (Huson and Bryant 2006). The data showed that the population of T. benhamiae is divided into four distinct subpopulations (designated S1-S4) and 32 genotypes. Numbers in parentheses indicate number of examined isolates from particular subpopulation. The subpopulation S1 was represented by highest number of isolates but at the same time had the lowest number of genotypes (clonal spreading) and is responsible for current outbreak of human and animal infections (mostly transmitted from guinea pigs) in Europe. Based on microsatellite and sequence data (not shown in figure) evidence, this clonal subpopulation is closely related to the subpopulation S2 represented exclusively by strains of North American origin (mostly infections in dogs) including the ex-type strain of T. benhamiae (marked with letter T)
is needed to confirm this assumption. Confusions are associated with phenotypes and genotypes recognized in the past among isolates identified as T. benhamiae. The isolates are usually designated as “white” or “yellow” phenotype based on macromorphology, and it was anticipated that they are connected with different genotypes (Symoens et al. 2013; Hiruma et al. 2015; Brasch et al. 2016). Apparently, the “white” phenotype which is predominant in Japan and in the USA and minority in Europe is in fact complex of several distinct taxonomic entities.
Random amplification of polymorphic DNA (RAPD) analysis (Kim et al. 2001) and PCR-RFLP targeting ITS rDNA (Heidemann et al. 2010) differentiated zoo- philic T. mentagrophytes and anthropophilic T. interdigitale. Some RAPD analyses were even useful for subtle subtyping of T. interdigitale (Kac et al. 1999; Leibner- Ciszak et al. 2010), but no correlation was detected between obtained profiles and geographic origin of strains. Poor reproducibility of the obtained profiles has reduced the interest in RAPD in favor of microsatellites and more reproducible methods in dermatophytes; but microsatellite markers have not been developed for T. mentagrophytes. PCR melting profile (PCR-MP) technique revealed seven genotypes within T. mentagrophytes and was able to distinguish zoophilic strains originating from Poland and Denmark (Leibner-Ciszak et al. 2010). Methods focusing on the variability in non-transcribed spacer (NTS) regions of rDNA have currently the highest discriminatory power among the methods applied on T. mentagrophytes. Southern blot hybridization-based RFLP analysis of NTS region was useful for subtyping of T. mentagrophytes and related T. interdigitale (23 subtypes among 60 isolates) (Mochizuki et al. 2003). Analysis of three individual subrepeat elements of the NTS identified 19 molecular types among 42 anthropophilic T. interdigitale strains (Jackson et al. 2006), and the method was later applied to 65 clinical strains isolated at one regional hospital in Japan with discrimination of 15 molecular types (Wakasa et al. 2010). Even higher resolution can be expected if the method was used to characterize closely related and sexually reproducing T. mentagrophytes.
Limited options are available for genotyping of T. verrucosum, T. erinacei, and T. equinum. It can be expected that at least some microsatellite markers designed for T. benhamiae will be useful for subtyping of T. verrucosum and T. erinacei due to close phylogenetic relatedness of these species (Fig. 3.3). Similarly, molecular subtyping based on single nucleotide polymorphisms (SNPs) developed for T. tonsurans (Abdel-Rahman et al. 2010) might work well in closely related T. equinum.
The establishment of global databases based on largely comparable data such as microsatellites, SNPs, and DNA sequences is desirable. Such databases would enable to understand global epidemiology of dermatophytes and monitor changes in genotype spectra on global and local scale. Although high throughput sequencing facilities are now widely available and increasingly used even in the epidemiology of fungal infections, this option has not yet been exploited in dermatophytes. SNP detection by using whole-genome sequence typing can be used, instead of MLST, to infer the genetic relatedness of fungal isolates. This ultimate approach will be certainly the method of choice in the future along with decreasing costs (Hadrich and Ranque 2015).
3.7