Hereditary and Congenital Myopathies
Stephanie J. Valberg
Mitochondrial Myopathy
A deficiency of complex I, the first step in the mitochondrial respiratory chain, has been identified in a young Arabian filly that was presented for veterinary attention with clinical signs similar to ER.267 In contrast to cases of ER, however, this horse showed no changes in serum CK following exercise.
A marked lactic acidosis developed even with light exercise, and maximum oxygen consumption was drastically reduced, resulting in marked exercise intolerance. Histopathologic evaluation of muscle biopsies showed an abnormal increase in mitochondrial density, and biochemical analyses revealed a complex I deficiency. The horse has shown slowly progressive signs of muscle atrophy but has otherwise remained healthy at rest.Glycogen Branching Enzyme Deficiency
Glycogen branching enzyme deficiency (GBED) is a glycogen storage disorder causing abortion, seizures, and muscle weakness in Quarter Horse-related breeds.268-270 It represents a separate glycogen storage disorder from PSSM. GBED is caused by a nonsense mutation in exon 1 of the GBE1 gene at codon 102, which introduces a premature stop codon.270 Nine percent of the breed is a carrier of this autosomal recessive mutation, and 3% of abortions are attributed to this disease in Quarter Horses.268 The prevalence of carriers is particularly high in pleasure horses, with 26% of pleasure horses carrying the GBE1 mutation.56 Most foals diagnosed with GBED have presented with hypothermia, weakness, and flexural deformities of all limbs at 1 day of age. Ventilatory failure may also be a presenting sign in addition to recurrent hypoglycemia and collapse. All foals have died from either euthanasia because of muscle
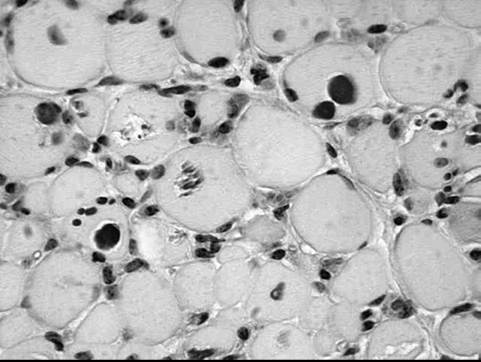
FIG.
42.20 Periodic acid-Schiff (PAS) stain of skeletal muscle from a foal with glycogen branching enzyme deficiency. Note the lack of normal diffuse dark staining for glycogen and the presence of globular and crystalline PAS-positive inclusions.weakness or suddenly because of apparent cardiac arrhythmia. Persistent leukopenia, intermittent hypoglycemia, and high serum CK (1000 to 15,000 U/L), AST, and γ-glutamyltransferase (GGT) activities are features of affected foals. Gross postmortem changes are not evident, and routine hematoxylin and eosin stains of tissues may be normal or show basophilic inclusions in skeletal muscle and cardiac tissues. Frozen sections of muscle, heart, and liver show a notable lack of normal PAS staining for glycogen, as well as abnormal PAS-positive globular or crystalline intracellular inclusions (Fig. 42.20). Branching enzyme activity is minimal in skeletal and cardiac muscle, as well as in liver. A diagnosis is best obtained by confirming the presence of the genetic mutation in tissue samples or by identifying typical PAS-positive inclusions in muscle or cardiac samples.269,270
Centronuclear Myopathy in a Foal
Progressive muscular weakness, lameness and atrophy including ventroflexion of the neck, ptosis, and lip droop have been described in an Arabian-cross foal that died by 5 months of age.271 Electromyography revealed fibrillation potentials, positive sharp waves, and complex repetitive discharges. Muscle biopsy findings were consistent with centronuclear myopathy that is associated with abnormal distributions of T-tubule and triad structures.
Phosphorylase Deficiency in Charolais Cattle
A deficiency of the enzyme myophosphorylase (McArdle's disease) has been identified in Charolais cattle in the United States and New Zealand and in sheep in Australia.272-275 Cattle show exercise intolerance and often collapse when forced to exercise. Serum CK is elevated, and severe rhabdomyolysis clinically resembling white muscle disease may be present.
Screening for myophosphorylase can be performed by histochemical staining for phosphorylase activity in frozen sections of muscle biopsies. Confirmation of the autosomal recessive disease is obtained by biochemical analyses or identification of the base pair sequence defect. In Charolais a C-to-T substitution, which changes an encoded arginine (CGG) to tryptophan (TGG) at codon 489, is present, and in sheep a splice-site mutation is present in intron 193. This disease should be considered a differential diagnosis for white muscle disease in animals that are found to have normal vitamin E and selenium status. An ovine form of myophosphorylase deficiency has also been described.276Pseudomyotonia in Cattle
Chianina and a Dutch Improved Red and White crossbreed heifer that have a form of pseudomyotonia characterized by delayed relaxation of muscle have been identified.277,278 Clinical signs include exercise-induced muscle stiffness with normal response to muscle percussion. In contrast to myotonia, electromyography and muscle histochemistry are normal. Missense mutations have been identified in the ATP2A1 gene encoding the sarcoplasmic reticulum SERCA1 protein.
Porcine RN(-) Glycogen Storage Disease
The RN(-) (rendement Napole) phenotype is common in Hampshire pigs. Clinically, pigs appear normal; however, the 70% increase in glycogen content in skeletal muscle causes poor meat content at slaughter. It is due to an autosomal dominant mutation in the protein kinase AMP-activated gamma 3 subunit gene (PRKAG3), which encodes the gamma 3 isoform of AMP-activated protein kinase.279 The mutation affects meat quality of pigs by increasing the glycogen content of muscle.