Macromineral and Bone Problems of Ruminants
Jesse Paul Goff
Introduction
Seven macrominerals, Na, K, Cl, Ca, Mg, P, and S, are considered essential for life. Inappropriate amounts of any of the macrominerals needed by ruminants can cause insufficiency and clinical disease including the “downer cow.” Ca disorders, such as milk fever, are not associated with insufficient diet Ca but rather an inability of the cow to use dietary Ca and bone mineral stores of Ca at the onset of lactation.
Na, K, Mg, and Cl are the major electrolytes of blood, extracellular, and intracellular fluids and influence blood volume, resting cell membrane potentials, and acid-base status. Many factors can contribute to the “downer cow syndrome” in addition to mineral challenges as outlined in Table 41.7. This chapter highlights the most common problems associated with each micromineral, focusing on identification of the problem, cause and prevention of the problem, and treatment options.Calcium
The Ca concentration in adult cow blood is normally 9 to 10 mg/dL (2.25 to 2.5 mM). Blood Ca concentration in calves younger than 4 months old will generally be a little higher, 9.8 to 11 mg/dL (2.2 to 2.75 mM), which may reflect the need for more Ca to promote calcification of new bone. About half of blood Ca is bound to proteins such as albumin and globulins, and a small portion (4% to 7%) is complexed to compounds such as citrate. The remaining Ca is in the blood in an ionized form at a concentration of 1 to 1.13 mM. This is the biologically active portion of Ca in the blood. It is the predominant form of Ca in the extracellular fluids bathing the tissues. The percentage of blood Ca that is ionized is affected by blood acidity; it varies from 42% to 43% of total Ca during compensated alkalosis and reaches 47% to 48% of total Ca during ■ TABLE 41.7 compensated acidosis. The 600-kg cow's blood normally contains approximately 3.25 g Ca, and the extracellular fluids of the cow contain a total of 10.5 to 11 g Ca (Fig.
41.19).Periparturient Dairy Cow Ca Metabolism
The onset of lactation imposes a large homeostatic challenge to the dairy cow. The day before calving, the 600-kg cow uses 7 to 8 g Ca for maintenance needs plus another 10 g Ca to allow fetal skeleton development, for a total of about 18 g Ca. To maintain Ca balance, 18 g Ca must be absorbed from the diet. Ca in dietary forages and concentrates are generally only about 30% available. If the forage and grains of the diet supply the Ca (no limestone or other more bioavailable Ca mineral is added to the diet) and the cow is consuming about 13 kg DM/day, the diet can be as low as 0.46% Ca and will meet the Ca demands of the cow.
At calving, the 10 g Ca needed for fetal skeletal development is replaced by a much larger Ca demand for colostrum and milk production. Holsteins produce an average of 7.5 kg first milk colostrum containing about 2.3 g Ca/kg. Harvesting that colostrum removes 17 g Ca from the blood. Within an hour of removal of the colostrum, the mammary gland will take up from the blood about three quarters of the Ca needed to form the transition milk (to be removed 12 hours later at the next milking).* 2 3 This is estimated to be another 13 g Ca. This brings the Ca lost from the blood to about 30 g Ca within a few hours of harvesting colostrum. The total Ca lost in the first day of lactation (milk Ca from first two milkings plus Ca sequestered in mammary for the next days milk) will approach 40 g Ca. Adding in the 8 g Ca maintenance requirement and the total amount of Ca leaving the blood on the day of calving approaches 46 to 48 g. This represents a 28- to 30-g Ca increase in net Ca that must enter the blood the day of calving compared with the day before calving. This represents removal of every g of Ca from the blood an additional eight to nine times during that first day compared with the day before calving. Amazingly, most cows successfully meet this challenge.
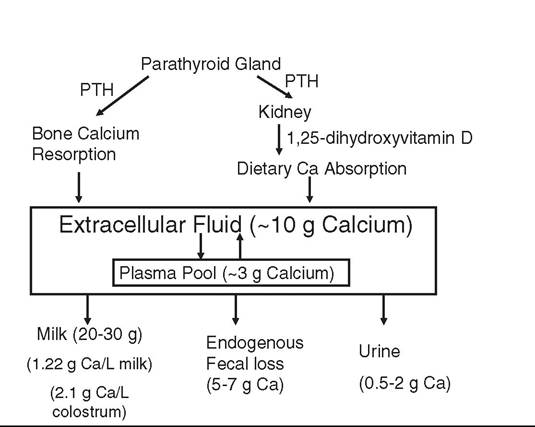
FIG.
41.19 Calcium homeostatic response of the 600-kg cow in early lactation to a decline in blood Ca concentration at the onset of lactation. PTH, Parathyroid hormone.Many cows, including nearly all first-calf heifers, will suffer only a small, temporary decline in blood Ca around the time of calving as Ca homeostasis mechanisms succeed in raising the blood Ca concentration back to normal. This is initiated by the parathyroid glands, which sense any decrease in ionized blood Ca concentration and secrete parathyroid hormone (PTH). G protein-coupled PTH receptors on bone osteoblast and osteocyte and kidney cells bind the PTH, resulting in an increase in intracellular cyclic AMP concentrations to initiate various actions within the target bone and kidney cells. PTH rapidly (within minutes) reduces urinary Ca loss, which can correct small Ca imbalances. However, because urinary Ca losses are typically below 0.5 g/day, it has a limited capacity to impact Ca balance at the onset of lactation. Continued secretion of PTH will stimulate osteocytic osteolysis, which involves moving Ca from the fluid surrounding each osteocyte sitting in its lacunae through the canalicular system and into the blood of the Haversian canal system in bone. This response is quite fast and moves about 9 g Ca into the blood of the typical cow within several hours. PTH will also stimulate renal conversion of vitamin D into the hormone, 1,25-dihydroxyvitamin D. This hormone acts on intestinal epithelial cells to stimulate active transepithelial movement of dietary Ca into the blood. This can bring large amounts of Ca into the blood, but it takes 24 to 36 hours to become fully active. Continued PTH secretion can also stimulate osteoblasts to secrete receptor activator of nuclear factor kappa-β ligand (RANKL). RANKL stimulates bone osteoclast recruitment and activation within trabecular and cortical bone. This has the potential to remove large amounts of Ca from the skeleton (over 1 kg Ca in the first 5 weeks of lactation) to help achieve Ca balance, but this process may take 48 to 72 hours to become fully active in older cows.
When operating properly, the cow experiences only a small decline in blood Ca concentration (class="lazyload" data-src="/files/uch_group31/uch_pgroup24/uch_uch7228/image/image652.jpg">FIG. 41.20 Parathyroid hormone (PTH) affects at the surface of target bone and kidney cells. (A) Under normal conditions, PTH released in response to hypocalcemia interacts with its receptor, located on the surface of bone and kidney cells, in a lock and key fashion. This stimulates G proteins and adenylate cyclase (adenylate cyclase complex) resulting in production of cyclic AMP, which acts as a second messenger within the cytosol of target cells. This initiates mechanisms such as bone Ca resorption and renal production of 1,25-dihydroxyvitamin D to restore blood Ca concentration to normal levels. (B) Alkalotic conditions induced by high-potassium diets induce a change in the shape of the PTH receptor protein so that it is less able to recognize and bind PTH, resulting in failure to activate the cell by producing cyclic AMP. (C) Mg is required for function of the adenylate cyclase complex. Hypomagnesemia reduces ability of PTH stimulated cells to produce cylic AMP, resulting in failure to activate the cell.
of cations and anions absorbed from the diet helps determine blood pH.3 Cows are typically in a state of compensated metabolic alkalosis as their diet consists of forages that are typically high in K+. Since K+ is absorbed from the diet with nearly 100% efficiency, forage K+ is strongly alkalinizing. Na+ in the diet or water is also highly alkalinizing as Na+ is also absorbed with nearly 100% efficiency. Ca++ and Mg++ cations can be present in diets in relatively high concentrations, but they are absorbed from the diet with much lower efficiency than K+ or Na+. Any absorbed Ca++ and Mg++ cations will also contribute to the alkalinity of the blood. Cows in a state of compensated metabolic alkalosis as a result of being fed a high K diet do not respond to PTH stimulation as well as cows placed in a state of compensated metabolic acidosis by addition of anions to their diet.
Metabolic alkalosis impairs bone Ca resorption. In addition, the ability of PTH to stimulate timely production of 1,25-dihydroxyvitamin D is impaired, reducing the utilization of dietary Ca.Adjusting Dietary Cation-Anion Difference to Reduce Alkalosis in the Cow
Since K is the primary mineral causing the metabolic alkalosis that interferes with PTH function, reducing diet K content is an important step in dry cow diet formulation. Mature forages contain less potassium than young forages, and forages grown where manure or potash has been applied to the soil will have high potassium content. Cool-season grasses and legumes will remove much more potassium from the soil than they need for growth, while warm-season grasses (including corn) do not. Forages harvested when the soil is moist (spring growth) generally have higher K content than forages grown during the drier months. By restricting K application to the soil, it is possible to avoid luxury consumption of K by legumes and cool-season grasses. Producers should also be aware that forages take up Cl from the soil and it is possible to find hays that are low K and high (1% to 1.2%) Cl. Producers should use the lowest dietary cation-anion difference (DCAD) forage possible, not simply the lowest K forage. Forages intended for the close-up dry cow should routinely be analyzed for both K and Cl by a wet chemistry method. Near-infrared determinations of forage mineral content are not currently useful.
Anions can be added to the diet to bring the cow into a state of compensated metabolic acidosis. Diet chloride anions are absorbed with nearly 100% efficiency. Sulfate anions can also help acidify the cow, but sulfate is not absorbed as well as chloride and has approximately 60% the acidifying activity of chloride. Although phosphate anions are acidifying, they should not be added to close-up dry cow rations as they actually impair Ca homeostasis (see Phosphorus later). When sufficient anions are added to the precalving diet, they induce a compensated metabolic acidosis in the cow, improving tissue sensitivity to PTH.
This restores the competency of Ca homeostatic mechanisms and facilitates a rapid return to normocalcemia after blood Ca concentration decreases at the onset of lactation.Blood pH is difficult and expensive to measure. Urine pH generally reflects blood pH and can be determined in cows on farm to determine the degree to which an anionic diet has placed the cow in a state of compensated metabolic acidosis. Urine pH can be checked 72 or more hours after a ration change. Urine samples should be free of feces and made on midstream collections to avoid alkalinity from vaginal secretions. In cows offered feed twice daily, the timing of the urine collection does not seem critical. In cows fed limited amounts of feed so as to achieve an empty bunk, the diurnal variation in urine pH can be a full pH unit. Checking urine pH routinely allows the farm to develop confidence that the DCAD is effective and safe. Published studies where anionic salts have improved periparturient Ca status have had an anion-supplemented group of cows with urine pH below 7.0 and usually closer to 6.0.4
Several DCAD equations have been developed to estimate the relative effect diet macrominerals and their charges might have on the animal's blood pH. DCAD mineral concentrations are expressed as mEq of the cations and anions-per-kg diet or per-100-g diet.
1. DCAD = (Na + K) - (Cl + S)
2. DCAD = (Na + K + 0.15 Ca + 0.15 Mg) - (Cl + 0.6 S +
0.5 P)
3. DCAD = (Na + K) - (Cl + 0.6 S)
The differences in the equations reflect whether or not all macrominerals were considered and whether efficiency of absorption of the mineral from the diet was considered. One meta-analysis of published studies on periparturient hypocalcemia found the equation DCAD = (Na + K) - (Cl + 0.6 S) had the strongest ability to predict urine pH and the strongest association with milk fever incidence. However, the equation DCAD = (Na + K) - (Cl + S) remains the most widely used equation.
Guidelines on the DCAD needed, expressed as (Na + K) - (Cl + S), to achieve different urine pH levels in cows are estimated below. DCAD of + 50 mEq/kg results in average urine pH of cows of approximaely 7.5. Urine pH will be around 7.0 when DCAD is approximately 0 mEq/kg and 6.3 when DCAD is approximately between -75 and -125 mEq/kg (Fig. 41.21). These are approximations, and it must be stressed that this particular DCAD equation overvalues the acidifying action of sulfate and ignores the alkalinizing action diet Ca and Mg have on blood and urine pH. Diets with anions added can be unpalatable and can depress DM intake (DMI). Commercial anion supplements that are more palatable than traditional ammonium, Ca, Mg, chloride, and sulfate salts have been developed. Even these products will depress DM intake once urine pH falls below about 5.9.
As DCAD decreases, the degree of hypocalcemia experienced by the periparturient cow will also generally decrease. There are, however, caveats to this premise. If the addition of anions to the diet fails to acidify the cow enough to cause urine pH to be less than 7.5, there will be no demonstrable improvement in periparturient Ca status. And if the DCAD is too low, the
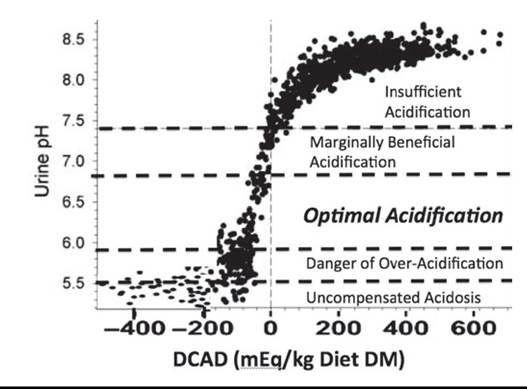
FIG. 41.21 Effect of dietary cation-anion difference (DCAD = [Na + K] - [Cl + S]) in mEq/kg on urine pH of individual cows. Zones of expected contribution to improvement of periparturient Ca status are depicted by horizontal dashed lines. The data on cows fed diets with DCAD below -250 mEq/kg are not well represented as feed intake declines precipitously in cows fed these diets. (Adapted from Charbonneau E, Pellerin D, Oetzel GR: Impact of lowering dietary cation-anion difference in nonlactating dairy cows: a meta-analysis. J Dairy Sci 89:537-548, 2006 and Constable PD, Gelfert CC, Furll M, Staufenbiel R, Stdmpfli HR: Application of strong ion difference theory to urine and the relationship between urine pH and net acid excretion in cattle. Am J Vet Res 70:915-925, 2009.)
cow is pushed into a state of uncompensated metabolic acidosis, which will greatly decrease DM consumption just before calving (see Fig. 41.21). As DCAD is reduced, the net amount of acid excreted into the urine increases until urine pH reaches about 6.3. When urine pH is below 6.3, the net acid excretion in urine is no longer well correlated to DCAD. Below 6.3, the kidney begins to also excrete ammonium cations into the urine, slowing a further decline in urine and blood pH. The ammonium arises from the combining of H+ with ammonia that diffuses into the tubular fluid as urine pH is reduced below 5.9. This ability of the kidney to neutralize the excess H+ allows the cow to remain in a state of compensated metabolic acidosis until urine pH reaches approximately 5.5. Urine pH below 5.3 is indicative of uncompensated metabolic acidosis, which will cause severe reductions in DM intake leading to ketosis, displaced abomasum, and immune suppression.2,5 There is evidence that dry cows with urine pH of 7.3 will be significantly less hypocalcemic than cows with urine pH of 8, but they will exhibit more hypocalcemia than cows with urine pH of 6.0. There is little evidence the degree of hypocalcemia experienced by cows with urine pH of 5.7 is less than in cows with urine pH of 6.3.
Another question revolves around the proper level of Ca to include in the diet of the cow fed anions. Anionic diets, ranging from 0.45% to 1.7% Ca, have been used successfully. Once the dietary Ca requirement of the cow has been met, the addition of Ca to precalving diets has little effect on periparturient Ca status if cows have been acidified to the same extent. However, diet Ca, especially when added as calcium carbonate, has a mild alkalinizing effect that will necessitate addition of more anions to achieve the same degree of urine acidification, which can increase the risk of DMI depression. The author prefers that prepartum diet Ca matches lactation diet Ca (0.8% to 1.1% Ca), with all of the Ca coming from forages and anion supplements.
Hypomagnesemia
A second common cause of hypocalcemia and milk fever in the periparturient cow is hypomagnesemia. Very low blood magnesium concentration (Lower doses are not effective and may actually induce milk fever because the high levels of 25-hydroxyvitamin D suppress PTH secretion and renal synthesis of endogenous 1,25-dihydroxyvitamin D. Recent studies using administration of the vitamin D metabolite, 25-hydroxyvitamin D, have not found that this compound can reduce the incidence of hypocalcemia, though its shorter half-life may make it less toxic than vitamin D.
Treatment with the hormone 1,25-dihydroxyvitamin D and its analogs can be an effective means of preventing hypocalcemia. Problems with cost, timing of administration and withdrawal from treatment, and lack of FDA approval have not made these treatments practical.
Synthetic PTH peptides given daily before calving can also effectively prevent milk fever, but no commercial product is currently available. Prolonged exposure of the bone and kidney cells to the high levels of PTH overcomes tissue resistance to PTH.
Other Milk Fever Risk Factors
Heifers rarely develop clinical milk fever, though 5% to 25% of them may experience subclinical hypocalcemia, with blood Ca falling below 8 mg/dL (Hypomagnesemia is a common problem only in ruminants.
Cow plasma Mg concentration is normally between 1.8 and 2.4 mg/dL (0.75 and 1 mM). Normal plasma Mg concentration in sheep is between 2.2 and 2.8 mg/dL (0.90 and 1.15 mM). The kidneys play a key role in maintaining Mg homeostasis by excreting excess absorbed Mg. The renal threshold for Mg (i.e., the plasma Mg concentration at which all Mg filtered across the glomerulus is reabsorbed is 1.8 mg/dL [0.75 mM] in the cow and 2.2 mg/dL [0.90 mM]) in sheep. Plasma Mg concentrations below these levels indicate dietary Mg absorption is not sufficient. Little or no Mg will be detected in urine.
Moderate hypomagnesemia between 1.1 and 1.8 mg/dL (0.5 and 0.75 mM) is associated with reduced feed intake, nervousness, and reduced milk fat and total milk production. This can be a chronic problem in some dairy herds that often goes unnoticed. It can also predispose these animals to hypocalcemia and milk fever as described earlier.
Role of Rumen in Hypomagnesemia
Mg is well absorbed from the small intestine of young calves and lambs. As the rumen and reticulum develop, these sites become the main, and perhaps only, sites for net Mg absorption. In adult ruminants the small intestine is a site of net secretion of Mg. Mg absorption from the rumen is dependent on the concentration of Mg in solution in the rumen fluid and integrity of the Mg transport mechanisms.
The soluble concentration of Mg in rumen fluid can be low for several reasons. Chief among these are low Mg content of forages and inadequate diet supplementation. Mg solubility also declines sharply as rumen pH rises above 6.5. Grazing animals tend to have higher rumen pH because of the stimulation of salivary buffer secretion. Forages can also contain organic compounds, such as unsaturated fatty acids, that are converted to tricarb allylate, which can form insoluble Mg salts in the rumen. When high grain rations are fed, rumen fluid pH is often below pH 6.5, increasing Mg solubility and thus availability.
Two mechanisms existing in rumen epithelium allow Mg to cross the apical membrane of the stratum granulosum cells in contact with the rumen fluid. One is a Na-linked transport mechanism, which relies on a high potential difference across the apical membrane to move Mg into the cell down its electrical gradient.8 When this transporter is fully functional, the ruminant can generally meet its Mg requirements with diets that are 0.2% to 0.25% Mg. Feeding ionophores (monensin, lasalocid) can improve activity of this Na-linked Mg transport system in the rumen, increasing Mg absorption efficiency about 10%. Lush high-moisture pastures increase the rate of passage of material, including Mg, from the rumen so that Mg may leave the rumen before it can be absorbed.
Unfortunately, the Na-linked Mg transporter is disrupted by high K and high ammonia/ammonium in the rumen fluids. High K concentration in the rumen fluid depolarizes the apical membrane of the rumen epithelium, reducing the electromotive potential needed to drive Mg thru the Mg channels in the apical membrane of stratum granulosum cells.8 Acute exposure to ammonia/ammonium similarly depolarizes the apical membrane of the stratum granulosum cells, inhibiting Mg transport by this route. Fortunately, a second Mg transport mechanism exists in the rumen epithelium that is not dependent on the potential difference across the apical membrane and therefore unaffected by diet K. However, this transporter is Mg concentration dependent (i.e., it is most effective when rumen fluid Mg ion concentration is high). Essentially, diet Mg has to be high enough to raise rumen fluid ionized Mg concentration above 3 to 4 mM. This may require the diet to be greater than 0.35% Mg, depending on the source of Mg and its solubility in rumen fluid. Mg content of the close-up dry cow ration and early lactation ration should be between 0.35% and 0.4% as insurance against the possibility that the active transport processes for Mg absorption are impaired.
Cows being fed a low DCAD diet to prevent milk fever and hypocalcemia are generally fed Mg as magnesium sulfate or magnesium chloride—both are highly available in the rumen and generally provide adequate amounts of Mg to use either of the rumen Mg transporters effectively. However, after calving, cows are placed on a lactation diet. Forages and magnesium oxide become the sources of Mg in these diets. Magnesium oxide (MgO) is used because it can also serve as a rumen fluid alkalinizer and help maintain rumen pH during lactation. Unfortunately, there is wide variability in availability of Mg from MgO and this often causes mild hypomagnesemia in cows during the first few days of lactation.
The reactivity of MgO with acetic acid provides a simple test of the relative availability of the MgO being considered for use in rations. Place 3 g of a MgO source in a container and slowly add 40 mL 5% acetic acid (white vinegar). Cap the container, shake well for 15 seconds, and let it sit. Shake again at the 15-minute mark and then check the pH at 30 minutes. Vinegar alone has a pH of 2.6 to 2.8. The best MgO sources will bring the pH up to 8.2 and the worst to just 3.8.2 Because pH is a log scale, this represents more than a 10,000-fold difference in the number of hydrogen ions neutralized. In an experiment with four cows with rumen fistulas, the solubility of MgO in vitro (tested in several ways) was found to parallel their solubility in the rumen and their urinary excretion—an indication that solubility predicted absorbability.9
Occurrence of Hypomagnesemic Tetany
Hypomagnesemic tetany is most often associated with beef and dairy cows and ewes in early lactation grazing lush pastures high in potassium and nitrogen and low in Mg and sodium. This is the most common situation, and it is often referred to as grass tetany, spring tetany, grass staggers, rye or wheat pasture poisoning and lactation tetany. Mg deficiency occurs most often in spring or fall when pastures are growing at maximal rates and is most common in lactating ruminants as milk production removes 0.15 g Mg from the blood for each liter of milk produced. Cool weather seems to play a factor as well, probably through its effects on plant Mg uptake, though there is some indication that the physiologic response of the cow to cool weather affects Mg status directly.
When plasma Mg levels fall below 0.5 mmol/L or 1.1 mg/dL, twitching is sometimes seen in the muscles of the face, shoulder, and flank. As hypomagnesemia progresses, tetanic spasms of the muscles become more common, which eventually cause the cow or ewe to stagger and fall. In most cases, the clinical signs of tetany occur only when hypomagnesemia has interfered with PTH secretion and function, causing secondary acute hypocalcemia.
Hypomagnesemia will slowly cause cerebrospinal fluid (CSF) Mg concentration to decline. Clonic convulsions quickly follow, with chomping of the jaws and frothy salivation. Affected animals lay with the head arched back and legs paddling. The heart rate can approach 150 bpm, and the heartbeat is often audible without a stethoscope. Respiratory rate approaches 60/min and the rectal temperature rises and can approach 40.5° C (105° F) as a result of excessive muscular activity. The eyelids flutter, and there is usually marked nystagmus. They may get up after several minutes and repeat these convulsive episodes several times before they finally die. Hypomagnesemic tetany in calves is clinically similar to that in adults and is often accompanied by moderate hypocalcemia.
Ewes are generally hypocalcemic and hypomagnesemic. Affected ewes are usually in the second to fourth weeks of lactation and are usually suckling more than one lamb. Affected ewes are generally depressed, stand with their heads down, and are reluctant to move. As hypomagnesemia and hypocalcemia progress, they suffer tetany and clonic convulsions just as in cattle. The clinical signs in goats are similar to those observed in cattle.
CSF Mg concentrations below 1 mg/dL (0.4 mM) are responsible for the clonic convulsions seen in animals with hypomagnesemic tetany. Unfortunately, blood samples obtained during or shortly after an episode of tetany may have nearnormal levels of Mg as a result of muscle damage and leakage of Mg from intracellular pools complicating diagnosis. CSF Mg concentration will remain low during tetany and also can be a reliable indicator of Mg status for up to 12 hours after death. Vitreous humor Mg concentrations below 0.4 mmol/L or 1 mg/dL are also found in animals with tetany and can be a reliable indicator for 24 to 48 hours after death provided that environmental temperatures have not exceeded 23° C. Aqueous humor has not proved a reliable sample.
Urine can be collected antemortem or postmortem. Low blood Mg concentration will not exceed the renal threshold for Mg, and as a result very little Mg will be in the urine. Urine Mg concentration below 1 mM is indicative of deficiency. Urine Mg concentration above 4.4 mM is considered normal. Urine Mg between 1 and 4.4 mM is likely insufficient Mg status.
Treatment of Hypomagnesemia
Animals exhibiting hypomagnesemic tetany need immediate treatment. This will require administering from 1.5 to 2.25 g Mg IV in the adult cow. They generally also need intravenous Ca. Most of the commercially available intravenous solutions used to treat the hypocalcemia of milk fever supply from 1.5 to 3 g Mg, usually as the chloride, borogluconate, or hypophosphite salts of Mg. Response to therapy can be disappointing, and success is related to the interval between onset of tetany and treatment. Cows should not be stimulated to rise for at least 30 minutes after treatment to avoid initiating tetany and convulsion. Cattle that will recover do so about an hour after treatment, which is the time it takes for CSF Mg concentration to return to normal. Many of these cows will relapse and require further treatment within 12 hours.
The rate of relapse can be reduced using orally administered Mg salts once the animal has regained good swallowing reflexes (to avoid aspiration pneumonia). Drenching or pumping into the rumen 100 g Mg oxide suspended in water can be effective. This provides about 52 g Mg to the animal. Adding 50 g Ca carbonate, 100 g dicalcium phosphate, and 50 g sodium chloride may enhance the effectiveness of the slurry, especially if hypocalcemia and hypophosphatemia accompany the hypomagnesemia. Alternatively, 200 to 400 mL of a 50% Mg sulfate solution can be administered by drench. Mg sulfate is more available for rumen absorption than Mg oxide. If hypomagnesemic tetany has occurred in one cow or ewe in a herd or flock, steps should be taken immediately to increase Mg intake in other members of the herd to prevent further losses. Getting an additional 10 to 15 g Mg into each pregnant cow, 20 g Mg into each lactating beef cow, and 30 g Mg into each lactating dairy cow each day, usually in a grain mix, will often prevent further hypomagnesemic tetany cases. Ewes and does can be treated with one eighth of the formulas mentioned earlier. The problem with these prevention schemes is getting the animal to actually consume the extra Mg, especially when working with animals at pasture. It can be added to a small amount of grain offered in the pasture. Mg sulfate and Mg chloride can be added to the water source, but this is only successful if the animals have no other water source to choose from.
Phosphorus
Traditionally, phosphorus in clinical medicine is abbreviated as P, although it should be understood by the reader that the biologically relevant form of phosphorus is actually inorganic phosphate, PO4-3, not elemental phosphorus. P is also a component of phospholipids, phosphoproteins, nucleic acids, and energy-transferring molecules such as ATP, but these are not detected as phosphate by the laboratory test used to measure blood inorganic phosphate. In the blood, phosphate ions are typically in the form of HPO4-2 or H2PO4-1. Moving phosphate between these two forms allows P to serve as an essential component of the acid-base buffer system of the body. P is second only to Ca as a major component of bone mineral, with the skeleton of a 600-kg cow containing about 3.9 kg P.
Phosphorus Homeostasis
Plasma P concentration is normally 4 to 8 mg/dL (1.3 to 2.6 mM). About 1.2 to 2.2 g P is present in the plasma inorganic P pool, and 5 to 8 g P is normally present in the extracellular P pool of a 600-kg cow. Intracellular P concentration is about 78 mg/dL (25 mM), and total body intracellular P content is about 185 g, with 5.5 to 6.5 g of P located within erythrocytes. Maintaining the extracellular P pool involves replacing extracellular P removed for bone and muscle growth, endogenous fecal loss, urinary P loss, and milk production with P absorbed from the diet or resorbed from bone. During late gestation, fetal skeletal development can withdraw up to 10 g P/day from the maternal P pools. About 0.3 g P is incorporated into each kg of body tissue (muscle) gained during growth of the animal. Production of milk removes about 0.9 g P from the extracellular pool/kg of milk produced. Salivary secretions remove between 30 and 90 g P from the extracellular P pool each day, though most of it is reabsorbed on reaching the small intestine. Factors affecting salivary phosphate secretion include the time spent ruminating (chewing activity) and the PTH status of the animal. PTH stimulates parotid salivary P secretion and can increase salivary phosphate concentrations twofold to threefold. Salivary phosphate secretions help buffer the rumen and supply rumen microbes with a readily available source of P necessary for cellulose digestion. Most of the salivary phosphate secreted is recovered by intestinal absorption. However, even on low-P diets a minimum of 5 g/day of secreted P is not recovered and is lost to feces. Urinary P loss is usually between 2 and 12 g/day. Bones of a 600-kg cow contain about 4 kg of P, some of which can be withdrawn and returned to the blood during osteoclastic resorption of bone.
Rumen microbes are able to digest phytic acid, so most of the phytate-bound P, the form of up to 70% of P in plants, is available for absorption in ruminants. P is primarily absorbed in the small intestine via an active transport process that is dependent on 1,25-dihydroxyvitamin D. This vitamin D hormone stimulates enterocytes to produce a Na+-coupled phosphate cotransporter protein in their apical membrane, allowing efficient absorption of diet P.
Homeostasis of P
Plasma P concentrations are generally well correlated with dietary P absorption. P absorbed in excess of needs is excreted in urine and saliva. Rising serum P concentration triggers the bone osteocytes to produce the hormone fibroblast growth factor 23 (FGF23). This hormone circulates to the kidney, where it inhibits the activity of the renal enzyme that produces the hormone 1,25-dihydroxyvitmin D, and stimulates the activity of enzymes that degrade 1,25-dihydroxyvitamin D (see Fig. 41.22). The lack of 1,25-dihydroxyvitamin D reduces the active transport of P across the intestinal epithelium, lowering blood P concentration. In addition, FGF-23 also acts on renal proximal tubule cells to block Na/PO4 cotransporters that reabsorb filtered phosphate from the tubular fluid, increasing urinary excretion of phosphate.10
Intestinal P absorption efficiency can be upregulated during periods of P deficiency as renal production of 1,25-dihydroxyvitamin D can be directly stimulated by very low plasma P, independent of PTH. However, the plasma P level must reach low levels (tend to become hypophos- phatemic. PTH could conceivably increase blood P concentration because it stimulates bone osteoclast mineral resorption. However, one reason the cow is hypocalcemic and secreting high levels of PTH is because the osteoclasts are not responding quickly enough to the PTH! PTH stimulates the kidney to produce 1,25-dihydroxyvitamin D, which should increase the efficiency of intestinal phosphate absorption. Complicating this further, a rise in blood P can trigger bone osteocytes to secrete FGF23 to shut off synthesis of 1,25-dihydroxyvitamin D, inhibiting intestinal absorption of Ca and P.
Acute Hypophosphatemia, Pregnancy Toxemia, and Downer Cows
Beef cows and ewes fed a diet marginal in P will have a chronic hypophosphatemia of 0.6 to 1.1 mmol/L or 2 to 3.5 mg/dL. In late gestation, plasma P can decline precipitously as the growth of the fetus accelerates and removes substantial amounts of P from the maternal circulation. These animals often become recumbent and are unable to rise, though they appear fairly alert and will often eat feed placed in front of them. Cows and ewes carrying twins are most often affected. Plasma P concentration in these recumbent animals is often less than 0.3 mmol/L or 1 mg/dL. The disease is often referred to as pregnancy toxemia and is usually complicated by concurrent hypocalcemia, hypomagnesemia, and in some cases hypoglycemia. Diets that are marginal in P are generally indicative of diets that are marginal in energy because grains are excellent sources of P.
At the onset of lactation in the dairy cow, the production of colostrum and milk draws large amounts of P out of the extracellular P pools. This alone will often cause an acute decline in plasma P levels. In addition, if the animal is also developing hypocalcemia, PTH will be secreted in large amounts, increasing urinary and salivary loss of P. In dairy cows, plasma P concentrations routinely fall below the normal range at parturition, and in cows with milk fever, plasma P concentrations are often between 0.4 and 0.75 mmol/L or 1.3 and 2.5 mg/dL. Plasma P concentrations usually increase rapidly following treatment of the hypocalcemic cow with intravenous Ca solutions. This rapid recovery is due to reduction in PTH secretion, which reduces urinary and salivary loss of P. Administration of Ca generally causes resumption of gastrointestinal motility, which allows absorption of dietary P and reabsorption of salivary P secretions that were sequestered within the rumen.
Some dairy cows developing acute hypocalcemia and hypophosphatemia do not spontaneously recover normal plasma P concentration. This is sometimes the case in cows that are classified as “downer cows.” This syndrome often begins as milk fever, but unlike the typical milk fever cow, plasma P remains low, often below 1 mg/dL (0.3 mM), despite successful treatment of the hypocalcemia. Protracted hypophosphatemia in these cows appears to be an important factor in the inability of these animals to rise to their feet, but why plasma P remains low is unclear. In some cases the inability to absorb the salivary phosphate is secondary to poor rumen motility, but not in all cases. Excessive cortisol secretion might be a factor in this syndrome as glucocorticoids can force extracellular P to move inside cells.
Treatment of these “downer cows” with phosphate (PO4-3)- containing solutions can effect a recovery in some animals provided they are treated before muscle and nerve damage have occurred. Unfortunately, many products suggested for intravenous use in treating this syndrome contain P as phosphite or phosphinic acid salts (PO2-2), which are biologically inactive. For proper oral treatment, the dose is 50 g P supplied in a 200-g monosodium phosphate drench. Intravenous treatment consists of 6 g P, supplied by 23 g monosodium phosphate, an amount found in many over-the-counter phosphate enema preparations sold in drugstores for human use. This author prefers to administer the phosphate dissolved in 1 L of saline. Oral treatment restores normal blood P more slowly than intravenous treatment, but the effect lasts longer. If depletion of intracellular P stores is involved in the downer syndrome, it seems likely that intravenous treatment alone will not supply enough P to replenish intracellular stores of P.
The hypophosphatemic downer cow syndrome does not appear to be caused by low P diets as affected cows are often receiving diets containing 0.4% dietary P. The best preventative measure seems to be to effective actions to avoid development of hypocalcemia. In the author's experience, hypophosphatemic downer cows are most commonly seen in the northern latitudes at the end of winter and early spring. Fluctuating temperatures caused by passing cold fronts may play a role.
Chronic P Deficiency
P deficiency is fairly common in ruminants grazing plants grown in arid climates or on tropical soils with poor soil P content or availability. While P content of the immature plant may be suitable (0.3% P-DM basis), the mature dry plants often contain less than 0.15% P. In more temperate areas, P deficiency can develop in animals grazing overly mature forages or crop residues, such as corn stalks. Sheep can, in some instances, be successfully raised on pasture that has been associated with P deficiency in cattle, suggesting that sheep are slightly more resistant to P deficiency syndromes than are cattle. This may simply reflect the higher intake/kg body weight of sheep and their habit of selecting the less mature plants, which are generally higher in P. With the exception of rickets and postparturient hemoglobinuria (discussed later), the clinical problems associated with P deficiency are general and nonspecific and can include an unthrifty appearance, reduced feed intake, pica, reduced rate of gain, and reduced milk production.
Impaired reproduction has often been attributed to “P deficiency.” However, in most cases cows develop P deficiency in situations that also result in concurrent energy deficiency. It is the negative energy balance that is the direct cause of reproductive failure. Unfortunately, the belief that “marginal” dietary P contributed to reproductive inefficiency has been used as justification for feeding diets that are much higher in P than is required. High-producing cows perform well in terms of milk production and fertility when fed diets containing just 0.38% P.11
Postparturient Hemoglobinuria
Intravascular hemolysis, anemia, and hemoglobinuria are occasionally reported during the first 6 weeks of lactation. Many, but not all, cows developing this syndrome are hypo- phosphatemic. Severe hypophosphatemia is postulated to depress the ability of erythrocytes to produce ATP because a key enzyme in glycolysis, glyceraldehyde-3-phosphate dehydrogenase, requires inorganic phosphate as a cofactor. Without sufficient ATP to power sodium pumps, the intracellular sodium concentration rises, the cells become more rigid and, as a result, rupture as they pass through the capillary beds.
However, hypophosphatemia alone is rarely sufficient cause for increased red blood cell fragility. Often these cows are on diets that are also deficient in selenium, copper, and energy, so attributing the cause to hypophosphatemia is likely too simplistic. Cows that have been treated for ketosis seem at greater risk of developing postparturient hemoglobinuria.
Electrolyte (Na, K, and Chloride)
Issues in Ruminants
Sodium and Chloride
Sodium is the major extracellular cation of body fluids and plays a crucial role in absorption of dietary sugars, amino acids, and water. Chloride is the major anion in the body, making up more than 60% of the total anion equivalents in the extracellular fluid. Chloride helps regulate osmotic pressure of blood. Chloride is the chief anion in gastric secretions and is responsible for the low pH in the lumen of the abomasum. Chloride is actively secreted into the intestinal lumen by crypt epithelial cells and acts as the driving force for Na movement into the lumen of the gut to satisfy needs of Na/hexose and Na/ amino acid symporters. Both Na and Cl always exist in a fully dissociated (ionized) state when in solution. Nerve and muscle resting membrane potentials are highly dependent on proper sodium and chloride concentrations. Most land vertebrates evolved without abundant diet Na. Therefore, they developed efficient and redundant mechanisms for Na absorption. Na and Cl transporters across the digestive tract epithelium are often coupled to maintain electroneutrality. Sodium and Cl can be absorbed from the rumen, small intestine, and large intestine. Nearly 100% of diet Na and more than 90% of diet Cl is absorbed.
Salt may be the only mineral compound that animals truly develop a craving for (perhaps phosphorus as well). Salt deficiency is often accompanied by observations of animals with pica, or abnormal appetite, with chewing or licking of wood, rocks, soil, urine, and bones being a common observation. Eventually osmotic and acid-base disturbances cause a reduction in appetite. Animals fail to grow, develop rough hair coats, and lactation comes to a halt. Long term, salt deficiency will cause death from dehydration. Nonlactating ruminants require 0.1% to 0.15% Na and 0.3% Cl. Lactation raises the requirement considerably to at least 0.4% Na and 0.5% Cl. For most species, the chloride requirement of the diet is adequately met by the combination of chloride from feedstuffs and from the salt added to meet the animal's sodium requirement.
Sodium and Chloride Toxicity
Sodium toxicity and chloride toxicity can occur separately. This is generally due to the effects these ions can have on acid-base physiology because, when fed separately, they can greatly affect the strong ion difference of the blood. However, when fed as sodium chloride the effects on acid-base physiology are negligible. Animals tolerate high levels of salt so long as they have free access to water to allow renal excretion of the salt. Salt will generally depress feed intake before the cow can ingest a toxic amount of salt. For most classes of cattle, voluntary intake of salt is up to 1.5 g salt/kg body weight per day. This amount of salt can be added to grain mixes to limit intake of grain/trace mineral/vitamin mixes fed ad libitum to animals at pasture.
Salt intoxication occurs in two phases. During the first phase, ingested Na and Cl are rapidly absorbed, causing a rapid rise in blood sodium and chloride concentrations, especially if water is restricted and prevents renal excretion of sodium. When plasma Na exceeds 160 mEq/L, water begins to move from the CSF into the plasma. If severe enough, intracellular water will be drawn from the cells in the brain causing cellular dehydration and brain shrinkage. As the brain shrinks away from the calvaria, the blood supply to brain cells can be disrupted, causing hemorrhage and thrombosis. If neuron dehydration is severe enough, convulsions and death follow. If not severe enough to kill the animal, a second phase of salt intoxication may occur over the next few days. In order to compensate for dehydration during systemic hypernatremia, brain cells try to increase the osmolarity of their intracellular spaces. This can only be partly accomplished by neuron uptake of sodium, chloride, and potassium from the CSF. However, the brain cells mostly rely on accumulation of organic osmoles such as taurine, glutamate, glutamine, and phosphocreatine within their intracellular fluids. If the hypernatremia is rapidly corrected, as might occur if water is suddenly offered to an animal that had limited access to water for a prolonged period, the osmolarity of the extracellular fluids would fall below that of the brain cell intracellular fluids. Water would tend to move across the blood-brain barrier and into the brain cells, causing them to swell. This occurs because the brain cells cannot remove the organic osmoles from the intracellular spaces as quickly as the kidneys (on ingestion of water) can correct extracellular sodium and chloride concentrations. Because the bones encasing the brain cannot expand to accommodate the edematous cells, pressure builds on the brain cells and causes necrosis of brain cells. This can cause convulsions and death. In veterinary medicine it is more common for animals to be observed in this second phase of salt intoxication. This syndrome is occasionally referred to as “water intoxication,” as the rapid restoration of normal blood osmolarity by water ingestion is often the factor precipitating clinical symptoms. Correction of severe salt toxicity or water deprivation should therefore occur over a period of 2 to 3 days so that the brain's adaptive mechanisms to prevent cellular dehydration and cerebral edema are not overwhelmed.
Potassium
Extracellular K concentration is normally 3.9 to 5.8 mmol/L and plays a vital role in osmotic equilibrium, resting membrane potential, and maintenance of acid-base balance. Intracellular K concentration is 150 to 160 mmol/L. Intracellular K is a cofactor of enzymes involved in protein synthesis and carbohydrate metabolism, and K plays a major role in intracellular osmotic and acid-base equilibrium. The ratio of intracellular-to-extracellular fluid K concentration is the main determinant of resting cell membrane potentials. Maintenance of normal blood K concentration requires a complex balancing act between K absorption from the diet and K removal from the blood.
Hypokalemia hyperpolarizes cells, causing resting membrane potential to be even more negative (-100 mv, inside negative, rather than the normal -90 mv), making it more difficult to reach the threshold for initiation of action potentials in nerve and muscle cells, resulting in paresis. If an action potential is successfully propagated down a motor nerve, hypokalemia, because it hyperpolarizes the resting cell membrane, will reduce the amount of acetylcholine released by the nerve ending at the motor end plate. This reduces the number of muscle fibers likely to contract, causing muscle weakness.
Moderate hypokalemia (plasma K from 2.5 to 4mM) is often due to prolonged inappetance, often secondary to other illnesses. Ketosis is commonly the factor precipitating the inappetance. Treatment of animals with steroids that have some mineralocorticoid activity has been shown to cause K concentration to fall low enough (below 2 mM) to cause muscle weakness and recumbency, causing the hypokalemic downer cow syndrome.
Hyperkalemia is most commonly encountered in ruminants secondary to the acidosis accompanying rumen lactic acidosis, ketosis, diarrhea, renal failure, and saliva loss during heat stress. Hyperkalemia initially reduces the K concentration gradient across cell membranes, making the resting membrane potential less negative (-70 rather than the normal -90 mv). Although this makes it somewhat easier to reach threshold potential, the depolarized cells have a lower ability to open Na channels at the start of the action potential. This decrease slows the inward sodium current, particularly in cardiac muscle. The slow entry of Na into the cell early in the action potential prolongs myocardial membrane depolarization and slows impulse conduction through the myocardium; as a result, an electrocardiogram reveals a prolonged QRS duration. Hyperkalemia can have drastic effects on cardiac function. Initial bradycardia due to depolarized cardiac muscle is followed by premature ventricular contractions due to failure of cardiac conduction giving rise to ectopic initiation of ventricular contraction. Finally, the heart stops in diastole.
Potassium Homeostasis
Cattle ordinarily consume diets that contain more than enough K to meet their tissue requirements. Because more than 90% of dietary K is absorbed across the intestinal tract, preventing an excessive increase in plasma K is a daily reality. The kidneys are expected to excrete any excess absorbed K. Renal K excretion is controlled by aldosterone, which enhances renal secretion of K in exchange for Na ions. High blood K concentration (hyperkalemia) can directly stimulate secretion of aldosterone by the adrenal glands. However, low plasma Na and low plasma volume are more potent triggers for aldosterone secretion. Because low plasma volume is the most potent stimulant of aldosterone secretion, it is possible to have scenarios where hypokalemia develops secondary to low plasma Na and low plasma volume. Aldosterone can also increase gastrointestinal secretion of K within saliva and pancreatic secretions, which can aid the kidneys in preventing hyperkalemia. It is important to point out that dietary K can enter the extracellular fluids rapidly following a meal, while the kidney will take several hours to excrete the excess K. Another mechanism is needed to prevent hyperkalemia following a meal. This mechanism involves shifting K from extracellular to intracellular fluids. Intracellular uptake of K is mediated primarily by insulin secreted in response to elevated plasma K concentration (and also in response to hyperglycemia or a rise in blood propionate). Insulin increases the activity of the Na-K ATPase pump, particularly in the liver and skeletal muscle, increasing uptake of K by these tissues. Ketosis can cause hypokalemia as a result of inappetance. And treatment of ketosis with intravenous glucose or oral gluconeogenic precursors, such as propylene glycol, will temporarily cause hypokalemia. These treatments cause a surge in blood insulin concentration, and the insulin will drive extracellular K into intracellular compartments.
β2-adrenergic catecholamines (i.e., epinephrine) can also drive K intracellularly. Epinephrine probably protects the animal from hyperkalemia during and following exercise. During exercise, K is lost from muscle during excitation-contraction of muscle. Stimulation of β2-catecholamine receptors stimulates Na-K ATPase to reduce extracellular K by driving K into muscle and liver.
Another confounding problem in understanding K metabolism is its subservient relationship with acid-base balance. Because disturbances in acid-base balance are debilitating or lethal, the maintenance of a stable pH of the extracellular fluids is of paramount importance. K can move between extracellular and intracellular fluid compartments. Movement of K from the extracellular fluids to the intracellular compartment reduces the strong ion difference of the blood, making the blood less alkaline (fewer H+). This is a compensatory mechanism that allows the animal to cope with alkalosis. Conversely, K leaves the intracellular fluids and enters the extracellular fluids during acidosis as a means of increasing the cation content of blood to raise the pH. (Some texts refer to this as K+-H+ exchange to move hydrogen ions in or out of cells.)
Blood K concentrations must be interpreted cautiously. Normal blood K concentration may not indicate normal intracellular stores of K, and abnormal blood K concentration does not necessarily indicate abnormal store or concentration of K inside cells.
Because dietary K is usually more than adequate, there is not an elaborate mechanism developed to avoid hypokalemia. Under most conditions hypokalemia is corrected by simply reducing aldosterone secretion. So long as the cow is eating, hypokalemia is generally easily avoided. However, if the cow is not eating, hypokalemia could quickly ensue, as illustrated by examining K balance in the cow.
K Balance in the Cow
In a 600-kg cow there will be approximately 1150 g K within the cells of the body and about 23 g K in all the extracellular fluids, with about 7.5 g K in the plasma pool. The maintenance requirement for K is the sum of the endogenous urinary K loss (amount of K lost in urine when cows are on a K-deficient diet, which is about 0.038 g K/kg body weight) plus endogenous fecal losses (≈6.1 g K is lost/kg DM ingested). Milk contains approximately 1.5 g K/kg, and fetal development in late gestation requires about 1 g K/day.12
If this cow is in early lactation and ingesting 14 kg DM/day, her maintenance requirement is about 108 g K/day. If her diet is 1.5% K, she is ingesting approximately 210 g K/day and about 189 g K is absorbed. If she is producing 15 kg milk the first day of lactation, she will use 22.5 g K for milk production. Of the 189 g K entering her body, 108 g K is used for maintenance and 22.5 g K is used for milk. This leaves 58.5 g excess K to be excreted in the urine if she is to avoid hyperkalemia.
If this cow suddenly goes off feed due to some secondary illness, K balance calculations would suggest this cow would rapidly go into negative K balance. Milk production would continue to withdraw 22.5 g K/day and the obligatory urinary K loss would be about 22 g/day for a net loss of 44.5 g K. There is some lag time between the cessation of aldosterone secretion and the cessation of K secretion by the kidneys, so the urinary loss may initially be higher than the obligatory K loss in urine. Considering that there is only about 23 g K in all the extracellular fluids, the cow could develop hypokalemia.
However, simply fasting a cow does not constitute enough of a challenge to K metabolism to cause the severe hypokalemia associated with downer cow syndrome. Holstein nonpregnant, nonlactating cows fasted for 48 hours had a decline in plasma K from 4.9 to 4.3 mmol/L. Plasma K of beef steers fasted for up to 4 days decreased from 4.1 to 3.7 mmol/L. A greater decline in blood K due to fasting is prevented by K entering the plasma pool from muscle. Hypokalemia alone can trigger a reduction in Na-K ATPase activity in muscle, leaving more K in extracellular fluids. However, reducing pump activity does not allow the muscle to replace K that chronically leaks from the muscle cell, especially during excitation of the muscle. Intracellular K concentration eventually falls, and intracellular Na concentration increases. This response is unique to muscle. Liver and other tissue intracellular K concentration does not decrease during hypokalemia.
During prolonged fasting, muscle is broken down and the muscle protein is converted to glucose. The K within the muscle cell is released during the gradual process of muscle breakdown that occurs with fasting. During the first weeks of lactation most dairy cows are in negative protein balance and, therefore, using large amounts of muscle protein to sustain milk production.
Reduced intracellular K concentration causes a loss of muscle strength. The loss of intracellular K diminishes the amount of Ca released from the sarcoplasmic reticulum during muscle activation and, as a result, the interaction between actin and myosin to cause muscle contraction is diminished. K ions must be bound to the Ca channel proteins within the sarcoplasmic reticulum if optimal Ca is to be released into the muscle cell cytosol following depolarization of the muscle cell membrane.
Hypokalemia and “Downer Cows”
K stored in muscle will generally prevent severe hypokalemia, if the animal is only off feed for a few days (before muscle is severely depleted of K). In most reports concerning clinical cases of hypokalemia and recumbency in cows, the plasma K concentration is less than 2.5 mmol/L. In many of the affected cows, plasma K concentration is less than 1.8 mmol/L. The degree of hypokalemia observed simply from inappetance of just 4 to 5 days is unlikely to be severe enough to cause flaccid paralysis in the cow. Though inappetance greatly reduces the amount of K entering extracellular pools, it seems other factors must be causing depletion of extracellular and intracellular K associated with the hypokalemic downer cow.
The possibilities causing low plasma K include exaggerated renal excretion of K and excessive uptake of K by cells. The possibilities causing intracellular K depletion include prolonged fasting and exaggerated renal excretion of K.
Factors Affecting Renal K Secretion
Excessive aldosterone secretion by the adrenal gland is relatively rare in cattle. However, drugs with glucocorticoid activity are often administered to cattle in early lactation as antiinflammatory agents or to stimulate gluconeogenesis in cows exhibiting ketosis. If those drugs also have mineralocorticoid activity, they will stimulate urinary K secretion. In the inappetant cow the muscles are releasing K to support normal blood K concentrations. If this K is being rapidly excreted by the kidney, the muscle K pool will be quickly depleted and severe hypokalemia can ensue. Isoflupredone acetate is sometimes administered to ketotic cows for its glucocorticoid activity, but it has enough mineralocorticoid activity to enhance renal K secretion. It should only be administered one time. Most hypokalemic downer cows had a history of being repeatedly injected with isoflupredone acetate over 2 to 3 days before the onset of recumbency.
Administration of diuretics, given primarily to treat udder edema, increases K excretion in the urine. Acetazolamide and the thiazide diuretics cause a large increase in urinary K loss. Furosemide, the diuretic most commonly used in cattle, increases K loss only moderately. Penicillin, aminoglycoside antibiotics, and gossypol (from cottonseed) also increase urinary K excretion to a minor extent. Though administration of diuretics and antibiotics have not been linked to hypokalemia and the downer cow syndrome in cattle, they have been implicated as the cause of cases of severe hypokalemia in man and companion animals.
FACTORS INCREASING TRANSFER OF K FROM EXTRACELLULAR TO INTRACELLULAR POOLS. Insulin causes a rapid uptake of K by muscle and liver. Administration of exogenous insulin was considered a factor contributing to hypokalemia in several cows developing severe hypokalemia. Endogenous insulin release occurring after intravenous administration of glucose or oral administration of glucose precursors such as glycerol, propionate, and propylene glycol will also shuttle K from extracellular to intracellular pools, but the effect is unlikely to be as long lived as with exogenous insulin.
β2-adrenergic stimulation will also drive K into muscle. Stress can elicit catecholamine release in cattle. Is stress of ketosis in early lactation a precipitating factor for hypokalemia? It does not seem likely since release of epinephrine or norepinephrine is not a major feature of ketosis in cows, as it is in humans. Fever may constitute a stressor that can elicit catecholamine release. A number of the cows described with the hypokalemic downer cow syndrome did exhibit a fever during the course of the disorder.
Systemic alkalosis will cause K to leave the extracellular fluids and could be a cause of hypokalemia. Hypochloremia, with or without alkalosis, is a common feature of the cases described by Peek and colleagues13. Metabolic alkalosis may have precipitated the hypokalemia, but because hypochloremia is a consistent observation in affected cows, it seems more likely that gastrointestinal ileus in the animals with severe hypokalemia caused sequestration of Cl within the abomasum. This alone will cause a metabolic alkalosis and acts to drive even more K intracellularly.
TREATMENT OF HYPOKALEMIC COW. Simply restoring plasma K is not enough. A true cure also requires restoring intracellular stores of K to optimize muscle function. Intravenous treatment is necessarily slow (to avoid hyperkalemia and heart stoppage) and should be limited to 0.5 mEq/kg/h. Oral therapy with KCl has proven a more effective therapy as enough K can be delivered to replace intracellular and extracellular K. It is also less expensive. Oral treatment consists of the administration of KCl at 100 to 150 g/dose given twice daily in several gallons of water for several days. Concomitant treatment with oral glucose precursors or intravenous dextrose to cause an insulin surge can help drive the absorbed K into the cells and will help treat the underlying ketosis present in most affected cows.14
If dehydration is present, plasma volume needs to be corrected first. Dehydration will cause aldosterone secretion, and much of the K absorbed across the intestine will be excreted in the urine without restoring intracellular pools. Rehydration with solutions that also provide Na will also help the cow restore normal acid-base balance.
SUBCLINICAL HYPOKALEMIA. Severe hypokalemia resulting in the down cow is a relatively rare phenomena, especially now that the role of repeated administration of steroids with mineralocorticoid activity is widely recognized and largely discontinued. Although intracellular stores of K have not been measured in affected cows, the course of the disease and poor response to intravenous K therapy alone suggest muscle depletion of K is a primary component of the disorder.
Moderate hypokalemia does not cause the flaccid paralysis associated with the hypokalemic downer cow syndrome. Moderate hypokalemia (2.2 to 3.9 mEq/L) is common in anorexic cattle, and severe hypokalemia is prevented by the release of K from intracellular stores in muscle.
If severe depletion of muscle K causes severe loss of muscle function and down cows, does moderate loss of K from muscle cause sluggish behavior in cows? Metabolism of K in cows is poorly researched because most diets provide more than ample supplies of K to the cow. However, the fresh cow is often faced with metabolic or infectious disease challenges that cause various degrees of anorexia. Would aggressive treatment of anorexic cows with oral KCl allow better muscle function in these cows, giving them the strength to fight for space at feed bunks, and restore them to productivity sooner?
Sulfur
Ruminants require sulfur and sulfate (SO4-2) to be in the diet to allow rumen bacteria to produce the sulfur containing amino acids, cysteine and methionine, and the B vitamins, thiamine and biotin, that the bacteria need to survive. The cow can make use of these bacterial amino acids and vitamins during digestion of the microbes once they exit the rumen. As a general rule, diets that are 0.20% to 0.23% S will satisfy the requirements of rumen bacteria. Diets consisting primarily of corn silage and corn grain risk S insufficiency. Organic sulfur in the form of the essential amino acid methionine and the B-vitamins is required by all animals. Sulfur and sulfate released during metabolism of sulfur containing amino acids can be used to produce compounds such as chondroitin sulfate and glutathione.
Of greater concern is the risk of S toxicity. In ruminants, S toxicosis presents as rapidly developing central nervous system symptoms, such as ataxia, blindness, and seizures, often followed by death. Brain lesions are most commonly described as polioencephalomalacia—swelling of the cerebrum, evidenced by flattened gyri and shallow sulci. The cerebral cortex may be thinned. The distribution is symmetric. Histologic section reveals a pale layer near the junction of the gray and white matter. The affected areas of the cortex will fluoresce under ultraviolet light.
Pathophysiology of S Toxicity in Ruminants
Some rumen microbes, particularly those found in cattle consuming high-concentrate diets, convert dietary sulfur to sulfide and sulfur dioxide, which can be reduced to hydrogen sulfide gas within the rumen. The hydrogen sulfide gas does not cross the rumen wall and enter the blood and would be relatively harmless if it stayed in the rumen. However, during the eructation process ruminants normally inhale a portion of the rumen gases into the lung. Hydrogen sulfide is absorbed across the lungs during eructation and causes symptoms of central nervous system disruption in the sheep. Hydrogen sulfide is a potent inhibitor of cytochrome C oxidase vital to mitochondrial respiration, and because the brain has a high energy requirement, hydrogen sulfide toxicity is associated with symptoms of central nervous system failure and brain lesions.
There is evidence that sulfur dioxide gas generated in the rumen can cleave thiamine to cause thiamine deficiency in the ruminant, leading to cerebrocortical necrosis. However, the lesions of polioencephalomalacia commonly described during sulfur intoxication of ruminants are similar, but not identical, to the cerebrocortical necrosis associated with thiamine deficiency. Further, most cases of polioencephalomalacia attributed to sulfur intoxication are not accompanied by decreased blood thiamine concentrations and do not respond to thiamine injection.15
Most reports of polioencephalomalacia are made in beef cattle or lambs on high-concentrate diets (90% grain) that exceed 0.4% S. Dairy cows, typically fed diets that are at least 40% forage, are rarely reported to suffer from polioencephalomalacia, despite the fact that addition of sulfate (in the form of anionic salts) to bring diet sulfur to 0.5% is commonly practiced as a means of reducing DCAD to control milk fever. Increased use of corn distiller’s grains has increased diet S intake, particularly in feedlot cattle. Sulfuric acid is commonly added to the distiller’s grains during processing to control pH of the fermentation, and it is used in cleaning the fermentation system. Distiller’s grains can be 0.6% to 0.8% S.
Water coming from wells can often contain high concentrations of sulfur, usually in the form of sulfate. Water quality reports typically express sulfur content of water in units of sulfate/L. Concentrations as high as 5000 mg sulfate/L are not uncommon. The maximal safe concentration of sulfate in water is 2500 mg sulfate (or 834 mg sulfur)∕L (0.08% S).
Other Possible Problems Associated With S
Many sulfur-containing compounds can be injurious to animals when ingested over the short or long term. Brassica plants (kale, cabbage, turnip, swedes) are included as a major part of the ration of ruminants in some parts of the world. These plants contain isothiocyanates that interfere with thyroperoxidase action in the thyroid gland, inhibiting thyroxine production and thus leading to hypothyroidism and goiter. Diet S can also interfere with bioavailability of diet trace minerals.
Bone Disease Associated With Ca, P, or Vitamin D InsufficienciesZExcess
Bone ash content varies somewhat with the bone examined but is generally 57% to 62% of bone dry weight. The ash will be 36% to 39% Ca, 16% to 18% P, and 5% to 7% Mg in adult animals. Ca and phosphate ions come together in a ratio of 10 Ca ions to 6 phosphate ions at the point of mineralization of the bone cartilage or osteoid matrix. Failure to supply adequate Ca, P, or vitamin D will prevent bone from mineralizing properly during bone growth in young animals and during remodeling of adult bone.
Growth Plates Allow Bones to Lengthen
Bone grows in length and width in young animals. Bone lengthens at the bone growth plates or physeal plates. A growth plate is a transverse disk of hyaline cartilage between the epiphysis and diaphysis. Chondrocytes within the growth plate become arranged into columns that run parallel to the length of the bone. The columns of chondrocytes are separated by longitudinal bars of hyaline cartilage matrix. Cells within each column are directly related to each other as they are the result of mitotic division within that column. Those chondrocytes within the growth plate that are the farthest from the diaphysis make up the zone of proliferation, where cells are rapidly dividing and extending the cartilage away from the diaphysis. The next visible stage within the growth plate is the zone of maturation or zone of hypertrophy. Here the chondrocytes have stopped dividing and have become enlarged. Glycogen accumulates within the cells, and their cytoplasm becomes highly vacuolated. The lacunae surrounding these chondrocytes expands, and only thin strands of cartilage matrix separate adjacent lacunae. Next, these small strands of remaining cartilaginous matrix begin to accumulate calcium phosphate deposits. This appears to be initiated by factors, such as type X collagen, produced by the hypertrophied cartilage cells. The actual process by which bone or cartilage matrix becomes mineralized is poorly understood. Some liken it to a solution that is supersaturated with calcium and phosphorus to which addition of a foreign substance initiates crystallization. In the case of bone the
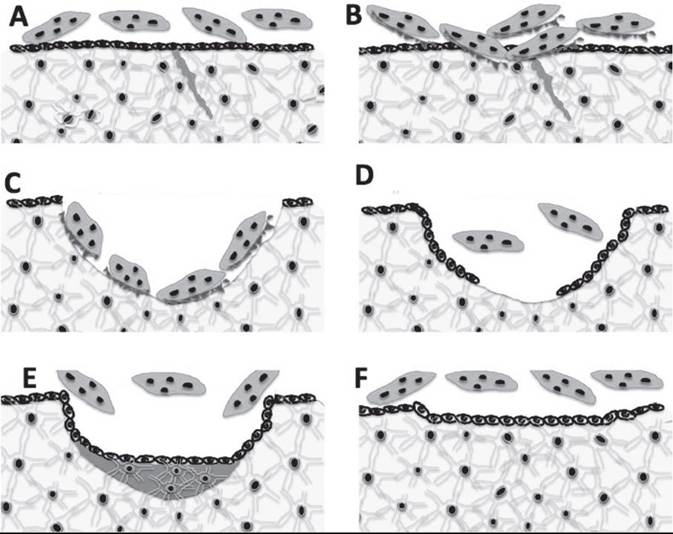
FIG. 41.23 Steps in trabecular bone remodeling. (A) Small area of bone with a microfracture destined for remodeling. Osteocytes in bone are connected to each other and to osteoblasts at endosteal surface by canaliculi. Inactive multinucleated osteoclasts rest outside the endosteum. (B) Activation. Osteoblasts retract from area of microfracture, and osteoclasts move into the area and develop a ruffled border. (C) Resorption of bone. Osteoclasts remove a shallow bowl approximately 50 μ deep into the trabecular bone. (D) Reversal. Osteoclasts move out of resorption lacunae, and osteoblasts move into lacunae from endosteum. (E) Bone matrix formation. Osteoblasts lay down unmineralized bone matrix. (F) Bone matrix mineralization nears completion. If blood Ca and phosphate are adequate, these minerals crystallize on γ-carboxylated glutamic acid sites of matrix proteins forming new bone to replace all the resorbed bone.
collagen fibers (or collagen in combination with glycoproteins or chondroitin sulfate) act as a nucleation catalyst, which transforms calcium and phosphate in solution in the tissue fluids into a solid mineral deposited on the collagen fibers. As the cartilage surrounding them calcifies, the hypertrophied chondrocytes undergo apoptosis or programmed cell death.
Finally, at the diaphyseal side of the growth plate, blood vessels invade the dead chondrocytes and calcified cartilage matrix and osteoprogenitor cells from the marrow spaces are brought into the calcified cartilage. As the lacunae are invaded, osteoblasts differentiate from the osteoprogenitor cells and form along the irregularly shaped bars of calcified cartilage that had once separated the columns of chondrocytes. A thin layer of bone matrix is deposited on the surface of the calcified cartilage. If conditions are favorable (adequate dietary calcium, phosphorus, and vitamin D), the new bone matrix quickly calcifies. This primary spongy bone area is often referred to as the metaphysis. As the growth process finishes, osteoclasts move into the metaphysis to reorganize and remodel the primary spongiosa to form true bone. Thus the rapidly dividing cells of the zone of proliferation continually advance the growth plate away from the diaphysis while osteoclasts and osteoblasts continually convert primary spongiosa to true bone at the diaphyseal side of the growth plate.
Bone Remodeling
Remodeling is necessary in large, long-lived animals to replace bone that is fatigued and accumulating microfractures. If bone remodeling came to a halt, the microfractures that accumulate would cause mechanical failure of the skeleton in about 2 years. Bone remodeling also serves another important function. It allows the skeleton to act as a repository of minerals by allowing transfer of calcium, phosphate, and other ions in and out of bone as needed to maintain electrolyte and acid/base balance. The hormones involved in calcium and phosphate homeostasis can greatly influence the rate and extent of bone remodeling. For example, PTH increases the number of bone sites undergoing remodeling.
Only tiny units of bone undergo remodeling at any one time. While bone remodeling in compact or cortical bone differs from that in trabecular bone, the same distinct steps— activation, resorption, reversal, and formation—occur during remodeling at both sites (Fig. 41.23).
1. Activation. This process converts a resting or inactive bone surface into a surface to be remodeled. How a particular section of bone is chosen for remodeling is not clear. The presence of microfractures in an area is one factor that marks an area for remodeling. This “selective” remodeling appears to be directed by a signal coming from the bone. When collagen fibrils are oriented correctly, there is a very minute electrical charge oriented along the fibrils. Microfractures upset the piezoelectric charge of that portion of bone and may initiate remodeling of that piece of bone. Deterioration of bone could also elicit secretion of autocrine or paracrine factors to initiate activation of bone remodeling. During activation, the bone lining osteoblasts retracts and shrinks in the area of bone to be remodeled, exposing the bone matrix. Multinucleated osteoclasts move onto this site, and a tight bond is formed between bone matrix and the osteoclasts. Endocrine factors such as PTH can increase the rate at which bone is remodeled, though this can sometimes lead to pathology as described later.
2. Resorption. The osteoclasts release acids and proteolytic enzymes from their ruffled border onto the bone surface, dissolving the organic matrix of the bone. The mineral crystals are dissolved, and the minerals along with the breakdown products of the organic matrix enter the osteoclast
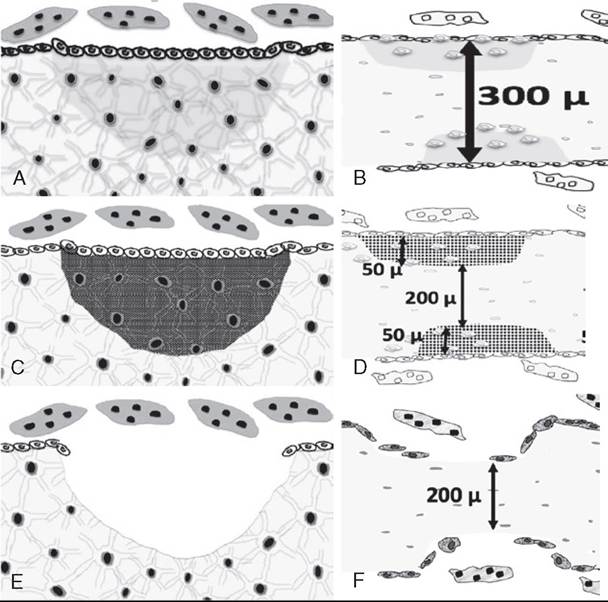
FIG. 41.24 Mineral-related problems during bone remodeling. (A) During normal bone remodeling, the bone removed from the Howship’s lacunae is completely replaced by newly formed bone. (B) A spicule of bone with remodeling sites on either side of the bone. The normal bone width and density of a spicule of bone will be the same before and after remodeling—in this case approximately 200 μ thick. (C) If the cow has not received adequate dietary phosphorus or is vitamin D deficient, the matrix is produced by the osteoblasts but fails to mineralize. (D) The remodeled area on each side of thespicule of bone area has 50 μ of rubbery matrix in each lacunae, resulting in osteomalacia, which allows bone to bend too easily. (E) If the cow receives too little dietary Ca or is in early lactation, the formation of new bone is uncoupled from the resorptive phase of bone remodeling. Neither bone matrix nor mineral is replaced. (F) The remodeled areas on each side of thespicule of bone area have not been replaced. This leads to a net loss of 50 μ of bone from each remodeling site, causing osteoporosis.
by diffusion or specific transport processes. They are then extruded into the extracellular fluid on the opposite side of the osteoclast.
The bulk of the bone resorbed to support Ca metabolism is trabecular bone. In trabecular bone the osteoclasts resorb bone until they have formed a saucer-shaped depression in the bone that is around 50 μ in depth at its center and 200 to 300 μ in diameter. In cortical or compact bone the osteoclasts drill out a cone-shaped section of bone to create a 200 μ-diameter tunnel of varying depth (10 to 100 μ). At this point the osteoclasts quit their bone resorbing activity and leave.
3. Reversal Phase. This phase signals the start of the rebuilding of bone within the resorption hollow, known as a Howship’s lacunae. Osteoblasts move from the bone surface down into the depths of the Howship’s lacunae. Several growth factors such as insulin-like growth factor (IGF)-II and transforming growth factor-β are thought to cause osteoblasts to migrate into the Howship’s lacunae to begin to replace the bone. Many other substances, including interleukin-1, platelet- derived growth factor, and IGF-I (somatomedin) are also considered candidates capable of initiating the reversal phase of remodeling.
4. Formation. Bone matrix formation begins with deposition of osteoid by the osteoblasts. This organic matrix consists primarily of type I collagen, proteoglycans, and smaller proteins typical of mature adult bone. In trabecular bone, the osteoid is laid down in discrete layers or lamellae about 3 mm thick with all the collagen fibrils within each lamellae oriented parallel to each other. In cortical bone the tunnel hollowed out by the osteoclasts is filled in from the outer circumference toward the center until only a small central canal containing blood vessels is left open. The collagen fibrils within different lamellae are oriented at an angle to the layer above or below, giving the bone added strength, an idea also used when lumber mills produce plywood.
Mineralization of the organic matrix is the final step in bone formation. It occurs after a time delay, which is about 20 days in man. Once triggered, the mineralization process is rather rapid. During the first few days of the process nearly three quarters of the total bone calcium will be deposited with the final 25% mineralization being completed months later. The total time for osteoblasts to complete the formation phase in a single Howship’s lacunae approximates 150 days (Fig. 41.24, A and B).
RICKETS. Rickets is a disease of young, growing animals in which the cartilaginous matrix at the growth plate fails to mineralize. The bones fail to grow in length, leading to short stature. The physes themselves are wide with irregular borders making joints enlarged. Rickets is a disorder of growing animals in which the newly formed osteoid and cartilage septa within the growth plate fail to mineralize. This is most often associated with vitamin D or dietary phosphorus deficiency. Rachitic animals will be hypophosphatemic and often hypocalcemic as well. Blood phosphorus concentration in young animals is generally higher than in adult animals—a reflection of their greater requirement for phosphorus to support mineralization of new growing bone. When blood phosphate concentration falls below the level required to support mineralization of the cartilage and primary spongiosa, the growth plate does not mineralize. Chondrocytes within the zone of proliferation continue to elongate the cartilage model in the growth plate. Vitamin D deficiency can reduce secretion of type X collagen by the chondrocytes within the growth plate, and this may prevent the programmed cell death of chondrocytes within the zone of provisional calcification. Since the cartilage does not calcify, blood vessels cannot move into the area to begin ossification and the physis elongates. The physis is more flexible and rubbery and gives the ends of bones an enlarged look, especially evident at the growth plates within the ribs.
Osteomalacia
During bone remodeling in adult animals, vitamin D or phosphorus deficiency causes blood phosphate concentration to fall below the level needed to mineralize newly formed bone matrix during the remodeling process. Nearly all steps in bone remodeling occur except for mineralization of the newly formed matrix. When vitamin D deficiency is the cause, the animals will be hypocalcemic as well. Serum concentration of 25-hydroxyvitamin D will be below 10 ng/mL (25 nM). The lack of phosphorus prevents mineralization but does not prevent normal bone matrix secretion by the osteoblasts (see Fig. 41.24, C and D). Pathologic changes occur much more slowly than in rickets. Over a prolonged period the bones become excessively flexible, leading to extensive joint pain in these animals. Rachitic bone and osteomalacic bone will have less bone ash per unit of bone volume or bone density. The ash will have below normal % P and % Ca content.
Osteoporosis Due to Dietary
Ca Insufficiency
Animals that are in a prolonged state of negative Ca balance develop osteoporosis. This bone disease can be observed whenever diet Ca is inadequate, while diet P and vitamin D are adequate. Low plasma Ca concentrations (arising from dietary Ca deficiency) can cause elevated secretion of PTH. The PTH causes increased activation of bone remodeling and increased bone resorption—there will be many more Howship’s lacunae formed in the bone. However, the prolonged secretion of high amounts of PTH prevents the reversal and bone formation steps of bone remodeling. Bone resorption and bone formation are no longer coupled as they are during normal bone remodeling. The result is bone that has many sites where osteoclasts have carved-out areas in the bone, giving it a moth-eaten appearance (Fig. 41.24, E and F). Bone density and ash content are greatly reduced, but the % Ca and % P in the ash are not greatly affected. The Ca content per unit of bone volume is greatly diminished. The bone does not bend as it does in rickets or osteomalacia. Instead, it becomes more brittle and is prone to fracture. Once diet Ca becomes adequate again, younger animals can restore the bone over time through renewed remodeling. Older cows may not live long enough to remodel all the sites damaged during prolonged dietary Ca deficiency.
Lactational Osteoporosis
During lactation, in virtually all species, there is an obligatory loss of bone mass from the skeleton as a result of uncoupling of bone formation from bone resorption. In this case the osteoclasts within an area of bone being remodeled resorb bone, to the normal 50-micron depth if diet Ca is adequate, or to even greater depths if the animal is also in severe negative Ca balance. However, osteoblast movement into the resorption cavity to replace resorbed bone is temporarily put on hold and no new bone is deposited. This occurs to some extent in all female mammals shortly after parturition, even when they are in positive Ca balance. However, the degree and duration of lactational osteoporosis can be magnified greatly by negative Ca balance. Once the animal has undergone the obligatory period of lactational osteoporotic bone loss (4 to 5 weeks in the cow), and if the animal is in positive Ca balance, the osteoblasts now come back into the resorption cavity and replace the lost bone. Lactational osteoporosis can help the female meet the Ca demands of lactation by uncoupling bone formation from bone resorption. In high-producing dairy cows, dietary Ca intake is generally inadequate to meet lactational Ca demands the first 4 to 6 weeks of lactation. The cow can lose as much as 13% of her skeletal mass during this period. The bone is replaced in later lactation when diet Ca intake allows the cow to enter a period of positive Ca balance.
Nutritional Secondary Hyperparathyroidism
A deficiency of dietary Ca or diets that are excessively high in phosphate can reduce the amount of Ca that can be absorbed across the intestine into the extracellular fluids. Diets severely deficient in Ca will not supply adequate Ca to replenish Ca lost from extracellular pools. As a result, blood Ca concentration decreases and PTH secretion increases. PTH causes the kidney to reduce urinary Ca loss and increase synthesis of 1,25-dihydroxyvitamin D. But if diet Ca is too low, increasing the efficiency of intestinal Ca absorption with 1,25-dihydroxyvitamin D cannot substantially increase the amount of Ca entering the extracellular pool of Ca. In addition, a high P diet raises blood phosphate concentration, stimulating bone osteocytes to produce FGF23. The FGF23 circulates to the kidney and blocks the 1α-hydroxylase enzyme responsible for producing 1,25-dihydroxyvitamin D. This also acts to block intestinal Ca transport. The only action of PTH that can improve blood Ca concentration under these situations is to stimulate bone Ca resorption by activating more sites of bone resorption. The high PTH, high P, high FGF23, and low 1,25-dihydroxyvitamin D content of blood causes the osteoblasts that move into the Howship’s lacunae to produce a more fibrous matrix than is normally produced during the bone formation phase of bone remodeling. The continued removal of bone without mineralization of the fibrous matrix results in fibrous osteodystrophy. The clinical signs of nutritional secondary hyperparathyroidism include hypertrophy of the parathyroid glands and cessation of growth in young animals. In young animals, the lack of Ca available for mineralization of bone matrix causes the growth plates to be soft, weak, and swollen. The cortices of the long bones are thin, and minor or major fractures are common. In older animals the bone of the mandible and maxilla become very malleable and teeth become loose, lending the moniker of “rubber jaw” and “bighead disease” to fibrous osteodystrophy of the bone.
Renal Secondary Hyperparathyroidism
This syndrome is due to renal failure and is occasionally observed in older cows suffering from pyelonephritis. A major function of the kidneys in most species is to remove excess phosphate from the circulation. As renal function is lost, phosphate is retained and hyperphosphatemia develops.
Hyperphosphatemia reduces the ionized Ca content of the blood. This is because Ca and phosphate ions normally exist in blood at concentrations that are slightly below the levels that would cause saturation of the fluids. The greatly elevated phosphate in blood of renal failure patients can exceed the equilibrium of Ca and phosphate in solution, causing a reduction of Ca from the blood. Lower blood Ca concentration increases PTH secretion. As phosphate builds up in the blood, it also stimulates bone osteocytes to produce the hormone FGF23, which blocks the activation of the 1α-hydroxylase that catalyzes conversion of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D within the kidney.10 Therefore, even though PTH should stimulate the remaining functional renal tissue to produce
1.25- dihydroxyvitamin D, this action of PTH is blocked by hyperphosphatemia.
As renal disease progresses, the amount of renal tissue available for production of 1,25-dyhydroxyvitamin D decreases, further compounding low blood concentrations of
1.25- dyhydroxyvitamin D. This reduces dietary Ca absorption and further depresses blood Ca concentration. This in turn stimulates increased PTH secretion and, resorbing bone Ca reserves, becomes the primary means of maintaining normocalcemia. Fibrous osteodystrophy follows the prolonged secretion of PTH when accompanied by elevated blood P and FGF23 levels.
As bone Ca is resorbed, more P is also resorbed. However, the loss of renal function prevents PTH from having its usual phosphaturic effect, exacerbating the hyperphosphatemia. A vicious cycle of higher blood P leading to increased PTH secretion and increased bone resorption ensues.
In principle, calcium deficiency differs from phosphorus deficiency in that normal osteoid is formed but fails to mineralize in phosphorus deficiency, while in calcium deficiency the normal osteoid is either not formed at all (osteoporosis if blood P and FGF23 are normal or low and blood 1,25-dihydroxyvitamin D is elevated) or replaced by fibrous tissue (fibrous osteodystrophy if blood P and FGF23 is high and 1,25-dihydroxyvitamin D is low). In adult vitamin D deficiency, it is common to see mixed lesions—areas of osteomalacia, osteoporosis, and fibrous osteodystrophy all in the same bone (Table 41.8).