Pseudorabies (Aujeszky's Disease, Mad Itch, Bulbar Paralysis)
Christine F. Berthelin-Baker • Lisle W. George
■ Definition and Etiology Pseudorabies is an acute, severe, and usually fatal encephalitis caused by an encapsulated DNA virus, Suid herpesvirus type 1, a member of the genus Varicel- lovirus, subfamily Alphaherpesvirinae, family Herpesviridae.1,2 This virus can infect the CNS and other organs (e.g., respiratory tract) of virtually all mammals except humans and most primates.
Domestic and feral swine are the natural hosts and may become latent carriers. The virus can also infect cattle, sheep, cats, dogs, goats, wildlife (e.g., raccoons, opossums, skunks, rodents), and, in rare cases, horses.3,4■ Epidemiology Pseudorabies has a worldwide distribution and, although cases occur only sporadically, is an economically important disease because of the regulatory quarantine and other restrictions imposed on animals from affected herds. This disease was endemic in the United States, but it has been successfully eradicated in domesticated swine. In the United States, all states are currently classified as free of pseudorabies in domesticated swine (OIE status V), although pseudorabies virus remains present in feral pigs in parts of the country.
The viral infection in cattle is perpetuated partly by the occurrence of latent infections in pigs, which can be recrudesced by stressful conditions.5 The virus is occasionally transmitted from swine into ruminants because of the proximity of the two species in many livestock operations.6,7 The natural routes of transmission include nose-to-nose, fecal-oral, and venereal.2,8 Indirect transmission usually occurs by inhalation of aerosolized virus. The virus may travel via aerosols up to 2 km (1.2 miles) in certain weather conditions and may survive up to 4 days in straw bedding. It is inactivated by drying, sunlight, and high temperatures (≥37° C).
Dead-end hosts such as dogs, cats, and wildlife can transmit the virus between farms, but aside from feral swine, these animals survive only 2 to 3 days after becoming infected.Latency of the viral infection in ruminants is not an important mechanism for perpetuation of an outbreak because most affected animals die within 48 hours of disease onset. In the few that survive longer, shedding in saliva occurs for up to 6 days after infection. The virus is unlikely to be transmitted directly from infected and uninfected cattle.7 Wild mammals such as raccoons may play a role in pseudorabies survival and transmission;9 rats, however, develop transient infections and do not appear to transmit or perpetuate the virus.10 The pseudorabies virus may survive in contaminated meat products for up to 7 weeks. The role of this prolonged survival in perpetuating outbreaks in ruminants is unknown.
■ Pathogenesis Ruminants are susceptible to pseudorabies infection after intradermal, subcutaneous, intranasal, or oral exposure to the virus. After subcutaneous, oral, or nasal infection, the virus spreads centripetally to the CNS by axonoplasmic transport. During the acute infection, the virus may be present in the nasal mucosa, secretions, and saliva.
■ Clinical Signs The incubation period ranges from 90 to 156 hours, and the illness may last 8 to 72 hours, although some affected animals have been found dead without having exhibited any clinical signs.6,11 The initial clinical sign in ruminants is usually acute and severe pruritus that induces self-trauma, dermal abrasions, swelling, and alopecia.11 Other signs may include fever, bellowing, bloat, feet stamping, excessive salivation, twitching, chewing of the tongue, ataxia, circling, nystagmus, and strabismus.12,13 Some affected animals exhibit aggression, but most become depressed. As the disease progresses, affected animals may exhibit hyperesthesia, tenesmus, excessive licking of the nostrils, continuous mastication, vocalization, coma, convulsions, and opisthotonos.
Most cases are fatal. The clinical signs of pseudorabies in cattle closely resemble those of rabies, polioencephalomalacia, salt poisoning, meningitis, lead poisoning, hypomagnesemia, and enterotoxemia.■ Clinical Pathology The pseudorabies virus can be isolated from the pharyngeal or nasal secretions of affected animals and can be easily cultured from infected nervous tissues. Strains of pseudorabies virus are antigenically distinct from other herpesviruses but share common antigens with the IBR virus. Heterospecific antibodies to the IBR virus may cross-neutralize the pseudorabies virus and confound serologic tests. For virus culture, tissues should be collected from sensory ganglia or the dorsal horn of the spinal cord. Segments serving the pruritic sites should be collected preferentially because these areas contain the highest concentration of virus.
■ Pathology Gross findings include excoriation, alopecia, edema, and hemorrhage at the pruritic site. The neuropathologic changes include nonsuppurative encephalitis, neuronal degeneration, and eosinophilic intranuclear (Cowdry type A) inclusion bodies. The lesions are most pronounced in the dorsal nerve rootlets and the dorsal horn.11
■ Treatment and Control There is no treatment for pseudorabies, which is usually fatal in livestock, but rare recoveries have been reported.13
The most effective method of preventing pseudorabies virus infection in ruminants is eliminating their exposure to swine. Contaminated pens can be disinfected with 10% sodium hypochlorite solution, quaternary ammonium compounds, tamed iodine, or phenolic compounds.14 At least 5 minutes of contact time should be allowed before the disinfectant is rinsed from the contaminated surfaces. Fumigation with formaldehyde for 6 hours effectively kills the virus, as does 360 minutes of contact with ultraviolet light.
Ovine Encephalomyelitis (Louping III)
■ Definition and Etiology Louping ill is an acute, fatal encephalomyelitis of sheep that occasionally infects humans, wild ruminants, horses, and cattle.1-3 The disease has been reported in England, Scotland, Ireland, Norway, Turkey, and Bulgaria.4-6 The etiologic agent of louping ill is a neurotropic, single-stranded RNA virus (flavivirus) that is transmitted primarily by the tick Ixodes ricinus, as well as by Rhipicepbalus appendiculatus, Ixodespersulcatus, and Haemaphysalis anatolicum.
Most outbreaks occur in swampy areas with dense populations of infected ticks and wild animals. Infection through blood contamination of hypodermic needles, other fomites, and blood products has been reported.7 Minor genetic variations of the viruses exist in different geographic areas, constituting four subtypes.8■ Epidemiology Louping ill affects principally yearling sheep in the spring, although lambs and adults can be affected. Maternal immunity wanes when lambs are approximately 3 months old. Infection develops weeks to months after sheep have been placed on pastures inhabited by infected ticks. In an outbreak, the prevalence of clinical louping ill is low, but the seroprevalence of antibodies in adult sheep in endemic areas is high, which indicates a continuous, low-level exposure to the virus, and many animals are affected without showing symptoms. Factors such as climate, tick population, and immune status of the flock all play a role in the severity of an outbreak.7 The attack rate may reach 60% of the population, but the mortality rate rarely exceeds 15%. Adults tend to be more heavily parasitized by the host ticks and thus play a major role in virus survival and transmission.9 Pigs, goats, cattle, horses, and red deer also become infected with the virus and can develop similar clinical disease.
In sylvatic cycles of virus transmission, viral amplification takes place through certain wild mammals and red grouse.1,10,11 The hare serves as an important source of virus for the red grouse.12,13 The red grouse is infected by ingesting, rather than by being bitten by, the infected tick.14
The high incidence of louping ill in the spring and summer months probably corresponds to the peak activity of ticks. The virus may persist for long periods in the arthropod vector, but it is unclear if transovarial transmission of the agent occurs. I. ricinus is a three-host tick with a life cycle of 3 years.
These ticks do not walk, and so dissemination requires animal transport. After a blood meal, the tick molts and then rests on vegetation until the next meal, approximately 12 months later. The activity of the ticks tends to increase greatly in the spring whenever the ambient temperatures are above 7° C (43° F).■ Pathogenesis Ticks become infected when feeding on a viremic host. The virus survives in the salivary glands of the tick and can overwinter there, being transmitted to a new host when the tick becomes active the following year.7 After inoculation into susceptible sheep, the louping ill virus migrates into the regional lymph nodes and spleen and then replicates. Viremia occurs 6 to 20 days after invasion into the lymphatic tissues.2 Viral replication in the brain causes nonsupportive inflammation and neuronal degeneration. Rapid antibody production is associated with recovery.15 Concomitant infection of sheep with the agent of tick-borne fever (Anaplasma phago- cytophilum) results in a greater level of viremia and increased mortality from the louping ill virus.16 The virus is transmissible in unpasteurized milk from sheep and goats.17
■ Clinical Signs In sheep, the initial clinical signs of louping ill include fever, anorexia, depression, constipation, and generalized muscular tremors. The ensuing signs are characteristic of CNS disease and include ataxia, conscious proprioceptive deficits, head tremors, hypermetria, and hyperexcitability. The hypermetria results in a characteristic “bunny-hopping gait,” which gives the disease its name (“loup” in vernacular Scottish means leap). Further progression of clinical signs is associated with cerebrocortical dysfunction; these signs include head pressing, hyperesthesia, recumbency, convulsions, coma, and death. Survivors have residual neurologic deficits. The duration of the illness is approximately 12 days.
■ Clinical Pathology Both hemagglutination inhibition and complement fixation tests can be used to detect virus in infected animals.7 High levels of virus-specific IgG and IgM can be detected in the CSF of affected animals.18 Viremia peaks at approximately 3 days after inoculation and disappears by 7 days.
Because animals are usually not viremic at the time the nervous system lesions develop, virus samples are collected most efficiently from the brain or spinal cord.■ Pathology Gross lesions are absent at necropsy. The histologic lesions of louping ill reflect meningeal inflammation with mononuclear cells.19,20 Microscopic lesions are most severe in the Purkinje cells, motor nuclei, and ventral horn cells. The forebrain is spared.21 Virus in tissue can be detected by virus isolation, immunohistochemistry, and RT-PCR.7,22,23
■ Treatment and Control There is no treatment for louping ill encephalitis, but supportive care should be provided.
A formalin-inactivated vaccine given to pregnant animals in the last trimester has been recommended for preventing louping ill encephalitis.24 A single dose of the vaccine provides responses that are protective for at least 2 years. Other methods of preventing the disease include frequent acaricidal dipping of sheep and clearing of pastures to reduce the population of infected intermediate hosts.
Louping ill has not been detected in the United States, but it is a reportable disease. Any suspicion of this disease should be reported to state and federal authorities.
■ Public Health Considerations The disease has zoonotic potential; infection of humans is rare but can occur through tick bites, unpasteurized infected sheep or goat milk, or fomites. Mild occupational infections have been noted in shepherds, veterinarians, and laboratory workers who cultivate the virus.
Rabies
Christine F. Berthelin-Baker • Lisle W. George
■ Definition and Etiology Infection with rabies virus results in severe and usually fatal neurologic disease. Most mammals are susceptible and are infected through bites from other animals during or near the clinical phase of illness. The rabies virus and other highly neurotropic rabies-related viruses belong to the genus Lyssavirus, family Rhabdoviridae (bulletshaped RNA viruses). Wildlife provides a natural reservoir for rabies, and each rabies strain is maintained in one or more particular reservoir hosts. Although they can readily cause rabies in other species, wildlife-adapted strains of rabies usually die out when passed into species to which they are not adapted. Other “rabies-related” lyssaviruses can cause a neurologic disease identical to rabies. They include the Lagos and Duvenhage viruses of bats in parts of Africa, the Mokola virus of rodents and shrews in Africa, and the lyssaviruses of European and Australian bats.1-3
■ Epidemiology Rabies is a reportable zoonosis that has been detected worldwide, with some exceptions (primarily islands). It currently does not occur in Sweden, Norway, the United Kingdom, Ireland, Australia, New Zealand, or Iceland.1 The disease is endemic in other parts of the world, including the United States, Canada, and Europe but remains epizootic in Central and South America, Africa, and parts of Asia, especially where a tropical climate and a high population of infected feral mammals enable viral propagation and transmis- sion.2,4 The rabies virus is shed in the saliva and does not survive when dried or exposed to ultraviolet light. The most common method of viral transmission to domestic animals is through the bite of an infected feral or wild mammal. Humans and laboratory animals have also developed fatal infections after respiratory exposure to viral aerosols. Low levels of rabies virus are also shed in the milk of infected animals and occasionally infect offspring nursing from infected dams.5,6 Laboratory animals, foxes, and skunks can also be infected experimentally by ingestion of infected tissues from animals with rabies.5,6 In the United States, survival of the virus in the wild may depend on cycling of infections in skunks, bats, raccoons, and foxes, which are also common sources of livestock infections.7,8
Certain strains of rabies virus have low virulence in skunks and bats; as a result, these animals are symptomless carriers serving as reservoirs. The rabies virus is also transmitted directly among bats through aerosols in caves. The bats may die off from rabies during periods of high viral contamination. In the United States the most common wild reservoirs are foxes in Alaska and along the border of Mexico, raccoons in the East, and skunks in the central and western regions.4 Mandatory vaccination of dogs has virtually eradicated canine rabies from industrialized countries, where rabies transmission from domestic dogs to livestock is rare.1,9 Island nations have remained free of rabies by imposing a 6-month quarantine on imported dogs and cats. In South America, rabies outbreaks in cattle often result from vampire bat bites, causing up to 50,000 cattle deaths annually. Human rabies cases may result from bites inflicted by infected pets, feral dogs and cats, bats, or wildlife. Cases resulting from
4 9 10 contact with infected cattle or horses are rare.4,9,10
■ Pathogenesis After subcutaneous or intradermal inoculation, the rabies virus replicates locally. Subsequently, it binds to the acetylcholine (and other) receptors of the peripheral nerves and then migrates to the CNS through peripheral nerves, spinal rootlets, and the spinal cord. After entry into the nerve cell rootlets, the virus travels to the brain along nerve tracts and into CSF. From there it spreads centrifugally along the rootlets of the cranial nerves to the salivary glands and the nasal epithelium. While inside cells, the rabies virus evades the immune system by blocking certain signaling proteins necessary for those cells to respond to interferons.11 The virus must replicate in the brain before it can reach the salivary and nasal secretions. Therefore whenever an animal bites a person, the attacking animal can always be euthanized and tested for the potential of rabies virus transmission, and it not necessary to wait for disease progression before testing an animal for rabies, as historically recommended when histologic examination for Negri bodies was the main diagnostic test for rabies. Negri bodies are aggregates of viral proteins and nucleic acids, most commonly observed on histologic examination of the hippocampus of rabies-infected animals. In humans, most cases become apparent within 1 to 3 months of infection, but a small proportion of cases have had incubation periods in excess of 6 months, extending to several years in rare cases.1 The rabies virus replicates in the cell bodies (gray matter) of the CNS. Dysfunction of these neurons results in behavioral changes and variable abnormalities of the cranial and the peripheral nerves, with multifocal loss of lower motor neuron and autonomic function. Death results from cardiorespiratory paralysis as the virus infects the medullary centers of the brainstem.
■ Clinical Signs Livestock cases of rabies have been reported mostly in cattle and occasionally in horses, sheep, and goats.3,4 Rabies should be included in the differential diagnosis of any unexplained acute, rapidly progressive intracranial disease, especially in the presence of autonomic instability, dysphagia, hydrophobia, paresis, or paresthesia.3,12 The incubation period ranges from a few days to 6 months, depending on the pathogenicity of the inoculum and the distance from the site of inoculation to the brain. The incubation periods are shortest in animals bitten on the head, whereas the longest incubations have been reported after bites on the extremities of the pelvic limbs.
The disease is usually fatal after a clinical course of 1 to 8 days. Occasionally, an affected animal is found dead without having exhibited any clinical signs previously. Early clinical signs of rabies in livestock may be nonspecific sensory and behavioral changes, including anorexia, restlessness, depression, separation from the herd, and mild ataxia. In ruminants, rumination may cease. Many affected horses appear colicky. Affected animals may show regional paresthesia with pruritus, creating excoriation and hair loss. As the disease progresses, affected animals may become hyperexcitable, fearful, or enraged (furious rabies) or mentally depressed (dumb rabies). Cattle rabies frequently manifests as dumb rabies with flaccid paraparesis, tetraparesis, and tetraplegia (paralytic rabies) as it progresses. Therefore the examiner should consider appropriate personal protective equipment (coveralls, boots, gloves, mask, and eye protection) when examining downer cattle in rabies-endemic areas.
In an experimental study, all cattle and sheep infected with the rabies virus exhibited excessive salivation and behavioral changes, and the majority also displayed tremors, bellowing, aggression, hyperesthesia, and pharyngeal paresis/paralysis.3 Tetraplegic animals may bellow and show frantic motor activity of the legs when stimulated. Animals with dumb rabies are depressed, inappetent, and often febrile (>39.4° C [103° F]), and they may exhibite head and neck drooping, ptosis, flaccid facial musculature, profuse salivation, and yawning. They may also demonstrate repeated nibbling motions with the lips. Other signs include tenesmus, paraphimosis, priapism, odontoprisis, head pressing, circling, basewide stance, difficulty rising, and falling episodes. Flaccidity of the tongue, tail, anus, and urinary bladder may also be evident, as may blindness, strabismus, and nystagmus.
The first clinical sign of paralytic rabies in horses may be an unexplained ataxia or shifting leg lameness, soon followed by paraparesis or paraplegia.13 Spinal reflexes and tone in the affected limbs may be decreased or absent. Most affected animals become recumbent in 3 to 5 days. Initially, the recumbent animal may be able to eat and drink with help, but it soon becomes anorectic and develops encephalopathic signs, followed by generalized seizures and coma14,15 (Fig. 35.11).
Regardless of the clinical manifestations, rabies is a rapidly progressive and fatal disease leading to cardiorespiratory failure, and death usually occurs within 10 days. In all disease forms in livestock, the clinical signs rapidly worsen over 1 to 3 days, ending in recumbency and coma. A pharyngeal-laryngeal paralysis may develop, resulting in stertorous breath sounds, inability to drink (hence the common name of hydrophobia), and accumulation of frothy saliva at the commissures of the lips.16
One summary of 21 cases of rabies in horses enumerated the frequency of particular clinical signs observed as the presenting complaint (Table 35.5). Rabies can be differentiated from other encephalitic disorders only by specific postmortem tests or virus isolation, which is usually not possible before death occurs. In horses, the clinical signs of rabies are often indistinguishable from other encephalopathies such as hepatoen- cephalopathy, leukoencephalomalacia, togaviral encephalitis, equine herpesvirus 1 (EHV-1), protozoal and other meningo- encephalomyelitides, and space-occupying masses. In ruminants, rabies may resemble pseudorabies, herpesvirus encephalitis, infectious thromboembolic meningoencephalitis, nervous ketosis, grass tetany, hypocalcemia, polioencephalomalacia, nervous coccidiosis, or even focal spinal cord or peripheral nerve diseases. Most horses with fatal encephalopathy die within 5 days of the onset of signs, but cases lasting as long as 14 days have occasionally been observed.17
■ TABLE 35.5
Clinical Signs of Rabies in Horses
| Clinical Signs | Frequency of Occurrence |
| Ataxia, paraplegia | 11/21 |
| Lameness | 5/21 |
| Pharyngeal paralysis | 2/21 |
| Recumbency | 21/21 |
| Colic | 2/21 |
| Hyperesthesia | 17/21 |
| Tail and anal paralysis | 12/21 |
| Fever | 11/21 |
From Green S, Smith LL, Vernau W, et al. 1992. Rabies in horses: 21 cases. J Am Vet Med Assoc 200:1133.
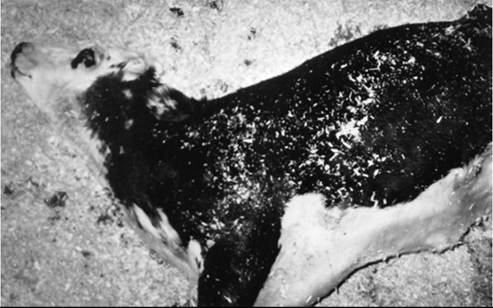
FIG. 35.11 Ten-month-old mixed-breed calf with terminal rabies virus encephalitis. The calf is comatose and showing opisthotonos.
■ Clinical Pathology There is no valid antemortem test to diagnose rabies in livestock. Rabies usually remains immunologically and serologically silent throughout the incubation and clinical phase of the illness. Clinicopathologic tests are not specific for rabies but may help rule out other, more common diseases. The CSF may be normal or may show moderate mononuclear pleocytosis (5 to 30 cells∕μL) and increased protein levels (60 to 200 mg∕dL), occasionally with neutrophils, eosinophils, or xanthochromia. Laboratory personnel should be warned that rabies is part of the differential diagnosis when CSF is submitted for analysis.
■ Pathology Rabies causes a nonsuppurative meningoen- cephalomyelitis. Microscopic changes include edema, meningeal congestion, focal areas of hemorrhage, perivascular cuffing, gliosis, neuronophagia, and neuronal degeneration. These changes are most severe in the dorsal root ganglia and can be observed in most CNS regions.
A definitive diagnosis of rabies may be based on the finding of Negri bodies on histologic examination of brain tissue (of note, these are not always present, especially in animals euthanized early in the disease course) or through a positive direct or indirect fluorescent antibody test conducted on fresh smears of CNS tissues (thalamus, hippocampus, brainstem, pons, medulla, and cerebellum).18-20 If results of these tests are negative or inconclusive and humans have been exposed, additional testing can be performed with intracerebral inoculation of mice with CNS tissue from the suspected case. The direct fluorescent antibody test on brain tissue has become the standard against which the value of other rabies tests is evaluated.18 In cases of human exposure, public health authorities should always be contacted before sample collection and submission. Most diagnostic pathologists accept the refrigerated head (or even whole body, for small animals and bats) or refrigerated brainstem and cerebellum collected through the foramen magnum in mature cattle and horses. Care should be taken to prevent any human exposure to body fluids and CNS tissue of any animal with rabies on the list of differential diagnoses, especially in areas where rabies is known to be present in any domestic or wildlife species; open wounds or small healing sores may be portals of entry for the rabies virus.
■ Treatment and Control There are no specific treatments for rabies, and once an animal is infected, the disease is usually fatal. Although widespread rabies vaccination of livestock may not be economically justified in low-incidence areas, vaccination of valuable livestock or animals that travel or are in regular contact with humans (e.g., at petting zoos or in animal shows) should be considered, particularly in or near rabies epizootic areas. A list of licensed rabies vaccines for the protection of animals against rabies is regularly updated by state veterinary authorities.21 Strict adherence to label directions for use of these vaccines is mandatory. In some jurisdictions, immunization of large animals is considered by regulatory authorities to be valid only if the vaccine is administered by a licensed veterinarian.
The disposal of livestock that have been bitten by rabid animals depends on the bitten animal's vaccination history, local and national regulations, and the animal's value. In the United States and Canada, the disease must be reported to the state or province/territory public health department. Management of exposed livestock must be coordinated with public health officials. Any livestock bitten by a wild animal should be considered to have been exposed to rabies, regardless of the availability of the biting animal for testing. If a bitten horse had been vaccinated before the bite, it should be revaccinated immediately and kept under observation for 90 days. Other exposed, unvaccinated animals with low economic value should be euthanized; if the animal is valuable, the bite wound may be washed with copious amounts of water and iodine or quaternary ammonium disinfectants, after which the animal should be quarantined for at least 6 months, and the brain should be examined if it dies.21 The prolonged quarantine period is based on case reports of prolonged incubation periods in certain species.1
Initial vaccination of animals shortly after rabies virus exposure is not recommended because it is less effective than prophylactic vaccination, and idiopathic responses to the vaccine may complicate interpretation of the animal's disease status after exposure.1 When human exposure to rabies virus is suspected, the biting animal must be euthanized and its brain examined for evidence of infection. A vaccinated or valuable animal may be quarantined for 6 weeks under veterinary supervision, and the animal must be euthanized and its brain examined if it develops clinical signs suggestive of rabies during that period. The World Health Organization (WHO) publishes and regularly reviews its recommendations for vaccination and prophylaxis in humans after actual or suspected exposure to the rabies virus.2
Control of rabies by immunization of susceptible wildlife through the use of baits containing modified live vaccines has 14 22 23
been successful in certain cases.1,4,, Such approaches may provide a means for large-scale immunization of wildlife or feral animal populations for eventual eradication of the disease.
Sporadic Bovine Encephalomyelitis (Buss Disease; Polyserositis; Chlamydophila pecorum Infection)
■ Definition and Etiology Sporadic bovine encephalomyelitis (SBE) is caused by Chlamydophila pecorum biotype 2, an organism previously named Chlamydia psittaci and Chlamydia pecorum.1 The disease was first reported in the United States in 1940.2 A similar but not identical viral disease occurs in cattle in Australia and has been named bovine ephemeral fever}-
■ Epidemiology Infected cattle shed the organism in urine, feces, nasal secretions, and milk. It can also be found in the feces of calves that are exposed to clinically affected herdmates but show no symptoms. The agent tends to remain endemic on a single farm, and sporadic outbreaks of disease may occur only on those premises. Outbreaks of SBE are rare, but the attack rate in an epizootic ranges from 5% among adults to 25% among calves. The mortality rate is highest in calves and approximates 30% for all age groups.6 The pattern of these outbreaks varies: from a few cases annually to acute, recurrent epizootics with high case-attack rates that subside after 3 to 4 weeks.
■ Pathogenesis The mode of transmission and the genesis of pathologic lesions are unknown in natural cases, but postmortem lesions indicate the role of hematogenous dissemination of the organism to the CNS and other tissues. The extracellular form, termed an elementary body, is metabolically inert and can be shed in the feces and urogenital secretions of multiple animal species.1 The pathogenesis of infection and invasion into the host's bloodstream is not well understood.
■ Clinical Signs Affected animals show signs of a multi- systemic disease. The initial clinical signs in cattle are fever (39° C to 41.5° C [102.1° F to 106.7° F]), diarrhea, anorexia, depression, and stiffness. Animals in the early stages may show signs of a respiratory disease characterized by nasal discharge, dyspnea, and cough. These animals occasionally have a pain response to percussion of the hoof, as well as swelling of the coronary band or polyarthritis and tenosynovitis.2 Auscultatory abnormalities may include high-pitched wheezes and crackles over the lung fields or pleural and pericardial friction rubs. Because of the fibrinous peritonitis and pleuritis, the clinical signs in some affected animals may resemble those of hardware disease. These animals may grunt or groan when sudden pressure is applied to the xiphoid region. Progression of the disease is related to development of the meningoencephalitis and is characterized by ataxia and conscious proprioceptive deficits, circling, head tilt, opisthotonos, hyperesthesia, stiff neck, convulsions, and coma. Affected animals may die after 4 to 10 days.
■ Clinical Pathology and Diagnosis The detection of reticulate bodies in the exudate cells of pleural and peritoneal effusions is highly suggestive of SBE. The agent can be most readily detected in clinical samples by PCR, although inoculation of laboratory animals may also be used.1 Serologic testing is difficult to interpret because antibodies to C. pecorum may cross-react with other Chlamydophila spp. on certain serologic tests.1
■ Pathology Growth of the organism in the arteriolar endothelium causes vasculitis, hemorrhage, edema, and accumulation of fluid in the body cavities. Diffuse fibrinous pleuritis, peritonitis, and meningitis are also present. Microscopic changes include perivascular mononuclear cell infiltration and neuronal degeneration.
■ Treatment and Control Early cases of SBE can be treated with oxytetracycline (20 mg/kg/day IV, SC, or IM for a minimum of 7 days). Efficacy is indicated by a reduction of fever within the first 24 hours of treatment.
There are no known effective control measures for the prevention of SBE. The chlamydial agent is susceptible to a number of disinfectants, including 2% sodium hydroxide (NaOH), 5% cresol, and 0.3% quaternary ammonium compounds.
Meningitis (Suppurative Meningitis; Bacterial Meningitis)
■ Definition, Etiology, and Epidemiology Bacterial meningitis is defined as inflammation of the meninges surrounding the brain or spinal cord, or both, that results from bacterial invasion of these tissues.1 If the infectious process extends into brain tissue, the condition is termed meningoencephalitis; if it extends into the ventricles of the brain, it is termed meningoventriculitis. Differentiation of meningitis from the other two conditions almost always necessitates advanced imaging techniques or histologic examination, but the conditions result in similar clinical signs.2 It is relatively rare in large animals. It is most often the result of hematogenous infection in neonates, as a consequence of septicemia with partial or complete failure of passive transfer of colostral antibodies or of septicemia resulting from translocation of invasive bacteria from the GI tract, respiratory tract, or umbilicus.2,3 Hematogenous infection of the meninges, brain, or both also occurs sporadically in adults; such events may be associated with septicemia from a primary focus of infection such as the udder, uterus, or GI tract. As with neonates, underlying immunodeficiency may predispose adult animals to this disease.4,5
Bacterial meningitis can also result from direct extension of infectious agents into the calvaria, such as extension of pyogenic infections into the calvaria from infected skull fractures,6,7 osteomyelitis from sinusitis or otitis media/interna,8 or osteonecrosis caused by thermal cauterization during dehorning (particularly in goat kids).9 Improper placement of trephination holes, cribriform plate fractures, and extension from infected coccygeal vertebrae are additional routes of meningeal infection. In horses, septic meningitis is a potential sequela to surgical removal of progressive ethmoidal hematomas and has also been reported as a consequence of local spread of infections of the paranasal sinuses, nasal cavity, periocular tissues, and submandibular lymph nodes.10
Many different bacteria have been isolated from the blood, meninges, brain, and spinal cord of affected large animals. In neonatal calves and foals, Gram-negative enteric bacteria such as Escherichia coli, Salmonella spp., and Klebsiella spp. are considered common isolates.1,2 Mycoplasma spp. may be isolated in goat kids. Histophilus somni is a potential cause of meningitis in juvenile cattle in feedlots and is reviewed later in this chapter, as are CNS infections caused by Listeria monocytogenes. A variety of Gram-positive and Gram-negative bacteria may be involved in adults, a fact that underscores the importance of cytologic examination, Gram staining, and aerobic and anaerobic culture of CSF samples obtained early in the clinical course of disease.1,2,11
■ Pathogenesis Bacterial turnover leads to liberation of exotoxins and endotoxin, which, in concert with various inflammatory mediators, including cytokines, damage the neural parenchyma. Vascular sequelae to infection may include thrombotic or hemorrhagic infarctions. After several days, inflammation of the arachnoid trabeculae and choroid plexus can result in decreased CSF absorption and hypertensive hydrocephalus.1,3
■ Clinical Signs The earliest clinical signs of meningitis are often nonspecific and may reflect the signs induced by a primary infection site (e.g., diarrhea in neonates with invasive bacterial enterocolitis). Fever is common, as is anorexia and loss of a coordinated suckle in a neonate.3 Reluctance to lift or bend the neck may be evident, and manipulations by the examiner may induce responses suggestive of hyperesthesia.2,3,12 Slight tactile stimulation of the skin may result in strong spasmodic extension of the limbs, fasciculations of the underlying musculature, or even generalized frantic motor activity. The patient's behavior may vary from extreme depression to hyperexcitability. A subtle intentional head tremor is often observed in affected foals. Dysfunction of one or more cranial nerves may result in facial muscular tremors, nystagmus, facial palsy, blindness, anisocoria, or strabismus. Progression of the clinical signs is associated with a decreased sensorium, propulsive walking, coma, seizures, and status epilepticus. Sequelae to septicemia, such as septic arthritis or polyarthritis, uveitis, panophthalmitis, hypopyon, cardiac murmurs secondary to valvular endocarditis, and pyuria/ proteinuria, may be detected as well.
The clinical signs of bacterial meningitis closely resemble those of viral encephalomyelitis caused by such agents as rabies, West Nile, and togavirus encephalomyelopathies (EEE, VEE, and WEE, described earlier in this chapter). Metabolic encephalopathies may also mimic bacterial meningitis including hypomagnesemia, hypocalcemia, hypoglycemia (which may occur simultaneously with septic meningitis in neonates), hepatic encephalopathy, renal encephalopathy, and encephalomalacias (e.g., salt poisoning, polioencephalomalacia). Congenital neurologic disorders of neonates may be considered; however, these disorders do not typically induce fever, inflammatory leukogram patterns, or inflammatory changes in the CSF.
■ Clinical Pathology Meningitis is diagnosed on the basis of examination of the CSF (for normal values, see Table 35.1). The CSF of animals with meningitis may be turbid and white to amber in color, may foam when shaken, and may clot. Xanthochromia may be observed in some specimens. The WBC counts in CSF of calves with purulent meningitis are typically greater than 100 neutrophils∕μL, and protein concentrations range from 20 to 270 mg/dL.3 The differential cell counts in the CSF are either predominantly neutrophilic or mononuclear. Because of bacterial consumption of glucose in the CSF, the concentration of glucose in the CSF is often less than 50% of the corresponding concentration in the serum. Gram staining of CSF should be conducted in all cases. Abnormalities in the blood are inconsistent and reflect secondary conditions such as septicemia, diarrhea, or concurrent fluid therapy. These changes could include leukopenia or leukocytosis, left shift, toxic changes in the neutrophils, hyperkalemia, respiratory acidosis, hypoglycemia, and hyponatremia or hypernatremia.
The numerous differential diagnoses discussed previously are distinguished from bacterial meningitis by compilation of data obtained from physical examination, serum immunoglobulin measurement, serum biochemistry analysis, complete blood cell count, and, of most importance, cytologic examination, Gram stain, and culture of CSE
PCR assays of CSF may be used to identify specific suspected organisms, as was done for diagnosis of meningitis caused by Borrelia burgdorferi in a horse.5
■ Pathology Gross lesions of primary infection in other organs or lesions suggestive of generalized sepsis may be apparent. When meningitis is secondary to trauma, the site of organism entry may be detectable. The meningeal vessels appear to be congested, and the meninges are swollen, opalescent, and petechiated. The CSF is cloudy or amber and may contain fibrin clots. Microscopic changes of CNS tissues include infiltration by neutrophils and lymphocytes, inflammation of the meningeal vessels, scattered hemorrhages, and bacterial colonies around the blood vessels of the meninges and the brain parenchyma.
■ Treatment and Control Early clinical suspicion of bacterial meningitis and rapid administration of appropriate antimicrobials are necessary to improve chances of survival and to minimize long-term neurologic impairment.1,2,11 In a human study of pneumococcal meningitis, a delay of antimicrobial treatment of more than 3 hours after hospital arrival was associated with a higher rate of morbidity 3 months after admission.13 Therefore it may be prudent to initiate appropriate antimicrobial therapy if the animal shows signs compatible with bacterial meningitis and then, if that diagnosis is subsequently refuted by ancillary diagnostic data, withdraw or modify antimicrobial therapy.
Treatment of bacterial meningitis is difficult, and the mortality rate is high, so treatment should be initiated only after owners are appropriately counseled. Antimicrobial sensitivity tests performed on isolates from CSF may provide valuable information on ideal drugs; however, drug selection must account for whether the drug will reach therapeutic concentrations in the CSF (discussed further later). Because prompt treatment of meningitis is critical, antibiotic therapy can initially be based on the Gram-staining characteristics of bacteria in CSF, when available, and on the initial 24-hour culture results, when possible. When expert laboratory cytologists are not immediately available, CSF can be placed into two microhematocrit tubes and centrifuged for 10 to 15 minutes. The tubes can then be scored with a file and broken at the interface of the fluid with the clay sealant, and the cell-rich fluid from the two ends of the broken tubes can be smeared for cytologic examination and Gram stain. In rabies-endemic areas, clinicians should take appropriate precautions when handling CSF.
Domestic animals have a well-defined blood-CSF barrier, and antibiotics that reach high plasma concentrations may not necessarily reach bactericidal concentrations in the brain. However, under conditions of local inflammation, this barrier becomes considerably more permeable by certain antimicrobials.1,11 As a general rule, in order to maximize distribution of antimicrobials across this barrier into infected meningeal or neural tissue, the highest dosage of antimicrobial established for the species in question should be used.1,11 Toxicity potential or excretory organ dysfunction may limit dose escalation of certain classes of drugs such as aminoglycosides. Intravenous administration is preferred in order to obtain peak blood concentrations and spur greater diffusion of drugs across this barrier. Because normal neural tissue lacks endogenous immune capacity, bactericidal antimicrobials have been considered superior to bacteriostatic drugs, although successful use of bacteriostatic drugs has been reported.1,11 The primary factors influencing CSF penetration of drugs in the healthy state are the lipid solubility (increased penetration with more lipid solubility) and molecular size (increased penetration with smaller size). However, the penetration capacity of a given antibiotic is not the sole determinant of appropriate antimicrobial selection because the blood-CSF barrier is made more permeable in bacterial meningitis. In fact, high-dose therapy with certain β-lactams, some with limited lipid solubility, either as monotherapy or in combination with antimicrobials of other classes, is currently recommended for many bacterial agents that cause meningitis.11
At present, use of all antimicrobial agents in treatment of bacterial meningitis in food animals and horses represents off-label use, and legal restrictions on use should be considered in all cases. Furthermore, the paucity of clinical reports and the variety of treatments used in those reports complicates the goal of making evidence-based decisions for treatment of this disease in horses and food animals. Therefore guidelines from published reviews of treatment in horses,1 cattle,3 and human medicine11 are presented here.
When possible, meningitis caused by Gram-negative enteric bacteria should be treated with specific third-generation (ceftazidime, ceftizoxime, ceftriaxone, cefoperazone, cefotaxime) or fourth-generation (cefepime) cephalosporins. For horses, ceftriaxone (50 mg/kg IV q6h) or cefotaxime (40 mg/kg IV q6h)1 have been used. Because of its high degree of protein binding in serum, ceftiofur (at 2 mg/kg IM) does not appear to spread into the CSF of normal horses.1 Its performance in penetration of the inflamed blood-CSF barrier remains to be determined. In the United States, off-label use of third- generation cephalosporins in food-producing animals is prohibited except under very specific circumstances, and thus current drug use guidelines within a given jurisdiction should be followed.
As an alternative, penicillin G sodium or potassium (20,000 to 40,000 IU/kg IV q6h) or sodium ampicillin (20 mg/kg IV q8h) may be used; success is considered to be highly dependent on the susceptibility of the causative organism, a parameter that is often unknown in the initial treatment decision process. If enteric organisms are suspected in a neonate, and if third- or fourth-generation cephalosporins are not available or are too costly for use, ampicillin is recommended; its Gram-negative spectrum is considered superior to that of penicillin G. Coadministration of an aminoglycoside can be considered for such neonates, with due consideration of nephrotoxicosis potential and, in animals intended for food, withdrawal time.1,11 Although aminoglycoside antimicrobials are highly effective against Gram-negative bacteria and are bactericidal, their efficacy in the treatment of purulent meningitis is diminished by their polar characteristics and low attainable CSF concentrations. Nonetheless, combination therapy consisting of a β-lactam and an aminoglycoside may provide effective broad-spectrum coverage11; the latter drug may at the very least reduce the circulating burden of Gram-negative bacteria in the bloodstream and elsewhere. Intrathecal administration of preservative-free aminoglycosides, although still used on occasion, has not been shown to produce tangible clinical benefit in comparison with intravenous use.11
Alternative treatments for meningitis caused by Gramnegative enteric bacteria, according to anecdotal data in horses only, include enrofloxacin (5 mg/kg, with due consideration of potential cartilage damage and reduction of seizure threshold,1 IV q12h), or trimethoprim-sulfonamide (5 mg/kg according to trimethoprim component, IV q12h).3 In horses, chloramphenicol (25 to 50 mg/kg PO q6h) may be considered for use.1 If β-lactam-resistant organisms are suspected, imipenem (10 to 20 mg/kg IV q6h) may be used, but its efficacy in treating meningitis in foals remains undetermined,1 and it carries the potential to lower the patient's seizure threshold.11 In the United States, off-label use of fluoroquinolones (e.g., enrofloxacin) and use of chloramphenicol in food-producing animals, as well as use of off-label use of sulfonamides in dairy cattle older than 20 months, are all prohibited by the Food and Drug Administration.
Meningitis caused by Gram-positive bacteria may be treated with high doses of intravenous penicillin, ampicillin, or the trimethoprim-sulfonamide combinations described earlier in accordance with the same provisos for food-producing animals. Third- and fourth-generation cephalosporins can be considered because these retain activity against certain Gram-positive organisms.1 For humans, moxifloxacin penetrates the blood-CSF barrier and has greater Gram-positive activity than earlier fluoroquinolones.11 Other antimicrobials showing promise against specific Gram-positive agents in human medicine include daptomycin and linezolid.11
Anaerobic infections are uncommon and probably result from penetrating or necrotizing wounds of the skull bones. Such infections in horses should be treated with metronidazole (15 to 25 mg/kg PO q6h)1; penicillin or ampicillin would be appropriate therapy in food-producing animals. Treatment of meningitis caused by Chlamydophila or Mycoplasma spp. has not been critically investigated in large animals, but the decision to treat with oxytetracycline, doxycycline, fluoroquinolones, or florfenicol depends on the animal class being treated. In general, treatment for 2 weeks is considered to be an empirical standard3; however, if aminoglycosides are included in the treatment regimen, the patient should be monitored carefully for nephrotoxicosis.
Adjunct therapy for bacterial meningitis includes analgesics, antiinflammatories, and antipyretic agents; fluid therapy; anticonvulsant therapy for seizures; protection against selfinduced trauma (including corneal trauma); and nutritional support. Maintenance of euvolemia and euglycemia is considered vital for treatment success.11 Hypoxemia and hypercapnia are deleterious and should be prevented whenever possible; mechanical ventilation of severely affected patients may be necessary. Osmotic diuretics such as mannitol or glycerol should be administered if CSF pressure, neurologic examination findings, or fundic examination findings are suggestive of cerebral edema. Use of narcotic agents for analgesia should be weighed against their potential to increase cerebral blood flow and therefore intracranial pressure. Although outcomes vary according to the causative organism, concurrent administration of dexamethasone may improve clinical outcome, although this remains controversial.11 NSAIDs may be a preferred choice if dexamethasone-related complications (e.g., laminitis in horses) must be prevented. Lmited food intake and the pain associated with this condition increase the potential for gastric ulceration, and prophylactic antiulcer medication should be considered in horses.
Treatments for seizures, cerebral edema, and inflammation are listed in Table 35.4.14-18 The maximum desirable trough concentration of phenobarbital is 40 μgfinL, and the minimum therapeutic level is 15 μg/mL.15,16 After initial sedation, longterm control of convulsions in horses can be maintained by oral phenytoin (2.8 to 16 mg/kg three times daily) or IV phenobarbital (11 mg/kg once daily).16-18 The trough plasma concentration of diphenylhydantoin should also be measured repeatedly during continuous drug therapy; the maximum desirable trough level is 10 to 20 μg/mL,17 and the optimum plasma concentration is 5 to 10 μgZkg. In critical care settings, constant-rate infusions of selected anticonvulsive drugs and drug combinations can be considered. The concentration of plasma immunoglobulins should be measured in neonatal patients with bacterial meningitis, and plasma should be administered if those values are subnormal for the species.
A guarded prognosis at best is warranted for most cases of bacterial meningitis. In a retrospective study of 32 calves with septic meningitis, only 5 (16%) survived until discharge, and all 5 subsequently died or had to be euthanized. In a case series in Spain, only 2 of 7 treated foals survived to discharge.19 The survival rate among adult horses may be higher. Early detection is considered pivotal for successful treatment; therefore a poor prognosis is warranted for animals with severe neurologic impairment. However, because concurrent metabolic derangements (e.g., hypoglycemia) may cloud the clinical picture, aggressive antimicrobial treatment—including achievement of euvolemia, euglycemia, and control of convulsions (if present) and normalization of acid-base, blood gas, and electrolyte status—may be attempted for valuable animals, with the prognosis refined on the basis of response to therapy.
Because cases of bacterial meningitis are usually secondary to other infections as discussed previously, prevention is centered on early detection and intervention for the primary disease. Ensuring adequate passive transfer of immunoglobulins in neonates is also important for reducing the risk of bacterial infection, septicemia, and meningitis.
Pituitary Abscesses result in exophthalmos. Extradural extension of the abscess along the brainstem causes loss of the cranial nerve function. Bradycardia may be caused by interference with diencephalic cardioacceleratory centers.
Animals aged 9 months to 12 years have been affected, but most cases occur between the ages of 2 and 5 years. Pituitary abscess appears to have a slightly increased prevalence among castrated and intact males.1 Pituitary abscess has been related to infections that develop in bulls after insertion of a nos e ring. Prophylactic administration of penicillin and attention to aseptic procedure during insertion of the ring can reduce disease incidence. Because of the high mortality rate associated with pituitary abscesses, treatment is not usually attempted.
Brain Abscesses
Streptococcus equi is the most common cause of brain abscesses in horses, probably because the occurrence of strangles in this species.1 T. pyogenes (formerly A. pyogenes) is a common cause of brain abscesses in cattle by means of extension of a sinus infection through the calvaria. Brain infections of cattle with Bacteroides spp. have also been reported.2 The neurologic dysfunction caused by brain abscesses typically has a slower onset and is more commonly asymmetric than that caused by meningitis. However, onset of signs can also be acute.1 Forebrain abscesses compress the cerebral cortex, causing a caudal displacement of the brain and functional loss of one or both occipital lobes, which may result in cortical blindness in one or both eyes. Because of the high proportion of crossed fibers in the optic nerve decussation, unilateral cortical abscesses may result in vision loss in the contralateral eye. Increased CNS compression by the mass may induce ipsilateral mydriasis caused by interference with the oculomotor nerve. Further increases in the size of the lesion cause more generalized cortical signs, including blindness, propulsive walking, circling, head tilt (toward the lesion side), depression, coma, head pressing, or sudden unexplained mania (Figs. 35.12 and 35.13). Abscesses at the base of the brain may cause additional abnormalities of cranial nerve function, including vestibular disease. Ophthalmoscopic examination may reveal papilledema in the ipsilateral eye.2 Advanced imaging studies such as CT and MRI, if available, may be helpful for antemortem diagnosis.3-5
In later stages of the illness, animals may assume lateral recumbency and display a decerebrate posture characterized by hypertonicity, hyperreflexia, opisthotonos, coma, and convulsions. At this stage the disease is difficult to differentiate
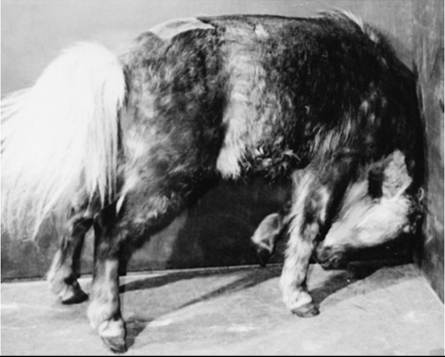
FIG. 35.12 Head pressing in a pony caused an abscess in the right cerebral hemisphere. (Courtesy Dr R.H. Whitlock.)
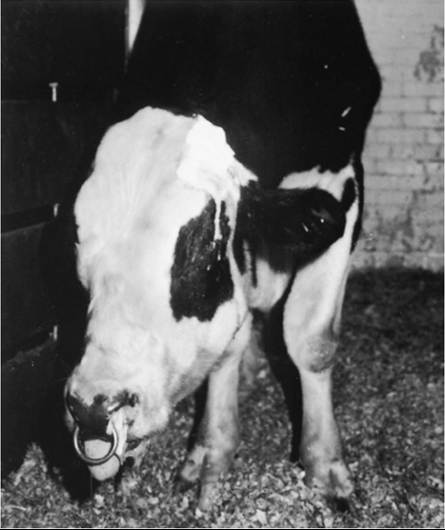
FIG. 35.13 Severely obtunded bull that developed a brain abscess from extension of a frontal sinusitis. The abscess extended to the base of the brainstem and affected cranial nerves V and XII, which resulted in dropped jaw and tongue paralysis. The bull was also severely depressed, blind, and ataxic and had facial analgesia.
from septic meningitis. CSF findings in animals with brain abscesses are variable, ranging from normal to very abnormal, with high protein concentration and marked pleocytosis.1,5,6 Treatment of brain abscess includes antibiotics and supportive care (see Meningitis section). In one clinical report, a brain abscess in a horse was localized with CT scanning and successfully drained through a craniotomy.7
Nervous Coccidiosis
■ Definition and Etiology Nervous coccidiosis is a neurologic syndrome of calves and yearling cattle, sheep, and goats that is associated with enteric infections by Eimeria spp. The condition is most often observed in western Canada and the northwestern United States and is particularly prevalent in feedlots. The incidence of nervous coccidiosis is highest in the winter months. In contrast to that of enteric coccidiosis, the rate of mortality from nervous coccidiosis is relatively high (≈70%).1 The pathogenesis of the encephalopathy may be related to the elaboration of a labile neurotoxin by the parasite.2 The clinical signs and history of nervous coccidiosis are similar to those of other neurologic diseases that affect the function of the cerebral cortex.
■ Epidemiology Nervous coccidiosis occurs most frequently in feeder cattle, but dairy and pastured beef calves, lambs, and kids are also be affected on occasion. In one survey, 0.3% of the calves were affected with the intestinal form of nervous coccidiosis. Nevertheless, outbreaks with a large percentage of calves developing CNS disease have been reported.3 In western Canada, CNS signs have been reported in 21% of cases in herd outbreaks of intestinal coccidiosis.4 Most cases occur in middle to late winter.
■ Pathogenesis The pathogenesis of the encephalopathy is unknown. The nervous form of coccidiosis cannot be transmitted to mice by injection of CSF from infected calves; however, a heat-labile neurotoxin has been identified in the serum of calves with nervous coccidiosis.2 The coccidia do not directly invade the CNS.
■ Clinical Signs The onset of the nervous system signs is usually preceded by diarrhea, tenesmus, and hematochezia. Some calves with severe diarrhea develop prolapsed rectums. Initial signs of CNS dysfunction include depression, incoordination, twitching, and hyperesthesia. As the clinical signs worsen, the animal becomes recumbent and develops numerous cerebrocor- tical signs, including opisthotonos, periodic tremors, horizontal nystagmus, frothing at the mouth, bellowing, snapping eyelids, and muscular fasciculations.3,5-7 Blindness is rare. Stimulation of the patient may precipitate a tonic-clonic seizure.3,5-7 Affected animals may die after 1 to 5 days of encephalopathy. Convulsive calves may regain consciousness but relapse days later.8
■ Clinical Pathology Fecal flotations from the patient and herdmates show a large burden of coccidial oocysts. Fecal egg counts of affected animals may range from 5000 to 4 million per gram of feces. To exclude the possibility of other neurologic diseases with similar clinical manifestations, blood should be collected for measurement of electrolytes (calcium, magnesium, potassium). The acid-base status, plasma glucose, and blood lead concentrations should be measured. Acute meningitis and salt poisoning may be ruled out by CSF analysis. The plasma vitamin A concentration should be measured in any animal that has not had exposure to green forage. Polioencephalomalacia, ethylene glycol poisoning, lead poisoning, rabies, pseudorabies, petroleum distillate poisoning, and clostridial enterotoxemia should be considered in the differential diagnosis.
■ Pathology No macroscopic lesions are observed in the CNS of calves with nervous coccidiosis. The microscopic lesions of the brain are mild and nonspecific and include edema, congestion, and occasional shrunken neurons. Parasitic invasion of the ileum, cecum, and colon results in lesions in these organs.
■ Treatment and Control Treatment should include supportive fluid, macromineral, and electrolyte therapy. The coccidial infection should be treated with sulfadimethoxine (55 mg/kg PO or IV, followed by 27.5 mg/kg PO or IV q24h). Alternatively, sulfamethazine (110 mg/kg PO q24h for 5 days, or 1 lb/100 gallons of drinking water) or amprolium (50 mg/ kg/day PO for 7 days) can be administered. Treatment administered via drinking water will be effective only if animals are willing and able to drink. Diazepam, sodium pentobarbital, or phenobarbital may be used to control tonic-clonic convulsions (see Table 35.4). Response to treatment is poor, and the case fatality rate can be high in calves that develop tonic-clonic seizures. Specific chemotherapeutic regimens and methods of preventing intestinal coccidiosis are described in Chapter 49.
Sporozoan Infections of Ruminants (Sarcocystis Infection)
■ Definition and Etiology The three recognized species of Sarcocystis that infect cattle—Sarcocystis cruzi, Sarcocystis hominis, and Sarcocystis hirsuta—are sporozoan parasites with definitive hosts of dogs, primates, and cats, respectively.1,2 For three other Sarcocystis species—Sarcocystis capricanus, Sarcocystis ovicanus, and Sarcocystis tenella—dogs are the definitive host, and secondary hosts include goats and sheep.3
When a carnivore ingests flesh from infected cattle, Sarcocystis spp. cysts in muscle are broken down by digestive enzymes, and motile bradyzoites are released. The bradyzoites infect intestinal mucosal cells and differentiate into sexual stages. The gametes fuse to form an oocyst, which is shed onto pastures as sporocysts. When eaten by a ruminant, the sporocysts hatch in the proximal small bowel and penetrate into the medium-sized mesenteric arteries, where they enter endothelial cells and form sporozoites. The sporozoites then mature in three successive waves. Each wave of development spreads downstream. The third-generation merozoites enter the soft tissues and encyst as sarcocysts. The total period of development in the ruminant requires 10 weeks. The life cycle is completed when a carnivore ingests uncooked meat containing viable sarcocysts. Disease may develop in cattle during the maturation of the cyst in the muscles, at approximately 9 to 11 weeks after infection.
■ Epidemiology Estimates of infection rates range from 70% to 98% in cattle in the United States.1 When infected flesh is eaten by carnivores, the encysted sporozoites complete their life cycle.1 The prepatent period of the parasite in the carnivore (primary host) ranges from 9 to 45 days. The primary host may shed the sporulated oocysts in the stool for as long as 2 months after a single infection. The oocysts withstand freezing but are rapidly killed by sunlight and drying.1 Reexposure of previously infected canids results in a large fecal output of sporocysts. Ingestion of approximately 250 g of infested meat by a dog can result in an output of 100 to 6000 sporocysts per gram of feces. Wild canids are even more susceptible than domestic dogs and may serve as a major mechanism for propagation of Sarcocystis spp. in range cattle in the western United States.
■ Pothogenesis The pathogenesis of Sarcocystis is poorly understood. Pathologic changes in the skin and muscle and in serum chemistry profile are speculated to result from a combination of parasite-directed immunologic responses and diffuse vasculitis. Abortions probably occur because of luteolysis that results from the increased concentrations of prostaglandin F2α, caused by vascular infection by the parasite.4
■ Clinical Signs Most cases of Sarcocystis spp. infestation are asymptomatic in both definitive and secondary hosts. However, if a large number of sporocysts are ingested by a nonimmune ruminant, clinical illness may develop. Clinical signs include fever (>39.5° C [103.5° F]), anorexia, weight loss, symmetric lameness, and diarrhea. Neurologic signs include ataxia, muscular weakness, tremors, hyperexcitability, hypersalivation, recumbency, tonic-clonic seizures, leg biting, blindness, opisthotonos, and nystagmus.5,6 Cattle may lose the hair of the tail switch (“rat tail”). Sheep may show wool break.1-3,5,6 Animals with chronic infections may develop edema of the limbs, poor weight gain, muscular atrophy, and pallor.1-3,5,6 Second-trimester abortions may occur in cattle and small ruminants beginning 28 days after ingestion of infectious sporocysts.7 The fetuses may appear either normal or autolyzed.
■ Clinical Pathology Serum activity of liver enzymes is increased. The packed cell volume and the serum protein concentration are often decreased. In the early stages of infection, there is a marked normocytic, normochromic anemia. The anemia is thought to be caused by extravascular hemolysis.8
Antibodies to solubilized freeze-dried Sarcocystis spp. antigens have been detected by indirect hemagglutination, ELISA, and an AGID test.9-11 IgM responses first occur by 3 to 4 weeks after infection and peak by 11 to 15 weeks.9-11 The concentrations of Sarcocystis-specific IgG begin to rise by 5 to 6 weeks after infection. Background titers of normal cattle range from 1 : 54 to 1 : 486, and titers of infected cattle often exceed 1 : 10,000.
■ Pathology The pathologic lesions in the CNS are similar for all species of sporozoans; they include granulomatous meningoencephalomyelitis, focal malacia, perivascular cuffing, neuronal degeneration, and gliosis. The changes are generally most severe in the cerebellum and midbrain but can occur anywhere in the CNS, including the spinal cord. The pathologic diagnosis is based on the finding of meronts and merozoites in the affected sections of neural tissue.12,13
Pathologic lesions elsewhere include hemorrhages on the sclera, serous surfaces, and muscles; fluid in the body cavities; and lymphadenopathy. The muscles have alternating light and dark stripes. Macroscopic changes may not be evident in animals with chronic sarcocystosis. If changes are not evident, postmortem diagnosis is based on the finding of intravascular schizonts or intramuscular hemorrhages without significant inflammation. Ultrastructural examination of affected areas of CNS reveals an intracellular colony with rosette orientation of agents in the cytoplasm of infected astrocytes.
■ Treatment and Control Treatment of infected sheep with salinomycin (1 to 2 mg/kg) has also been recommended.14 Amprolium (100 mg/kg once daily for 30 days) may reduce the severity of Sarcocystis spp. infection15 but may not completely eliminate the clinical disease.
The best method of controlling Sarcocystis spp. infection is to protect the food supply of ruminants. Scavenging of carcasses by carnivores should also be prevented by deep burial or incineration. Feed bunks should be kept clean and raised approximately 1 to 3 feet (30 to 90 cm) off the ground. All carnivorous pets that have access to the feed or pastures should be fed cooked meat or processed dry food. In range pasture situations, prophylactic feeding of monensin or elimination of predatory or scavenging carnivores may be necessary.
Neospora Infection of Cattle (Protozoal Abortion)
Neospora caninum is a protozoal parasite that causes disease primarily in dogs and cattle.1 The predominant clinical sign of Neospora caninum infection is a midterm to late-term abortion (3 to 8 months of gestation). Fetal lesions consist of focal nonsuppurative necrotizing encephalitis, nonsuppurative myocarditis and myositis, and mononuclear cell infiltrates disseminated in other tissues.2,3 Occasionally, stillborn calves are mummified. (See Chapter 43 for more on abortion.)
Nonfatal infection in the fetus results in neurologic dysfunction of the newborn. The clinical signs of neurologic disease vary because of the random and widespread distribution of the parasite within the CNS. Affected calves are often unable to stand and suckle and have abnormal spinal reflexes. Flexural contractions of the forelimbs, domed skull, and torticollis have also been reported in spontaneously occurring cases.4,5 Calves born alive are usually born with the CNS signs, which initially are mild but then worsen after birth. Pathologic lesions associated with the fetal infection include focal areas of brain discoloration, focal cavitation with cyst formation, and reduction of gray matter. Microscopic changes in the CNS of affected calves include nonsuppurative inflammation of the gray and white matter, focal lymphocytic meningitis, and neuronal necrosis. Changes in other tissues include nonsuppurative myocarditis, myositis, and hepatitis. Protozoa can be observed in microscopic sections of the stained tissues.
Neospora organisms have been isolated in pure form in cultured cells.6 Antibodies have been produced by intubation of laboratory animals, and the agent can be identified microscopically through immunoperoxidase staining on the fixed tissues. The CSF changes in affected calves range from normal to mild pleocytosis.7 Similar conditions have been described in sheep8,9 and goats.10,11
Babesia Encephalitis (Babesiosis; Piroplasmosis; Texas Cattle Fever; Tick Fever; Redwater)
Parasitemia of cattle caused by the protozoans Babesia bovis, Babesia argentina, and Babesia is usually subclinical, but results in devastating economic losses worldwide.1 The disease is transmitted to cattle by the cattle fever ticks Rbipicepbalus (Boopbilus) annulatus, Rbipicepbalus microplus, and Rbipicepbalus decoloratus. Babesiosis occurs in the Americas, Europe, Africa, Asia, and Australia. Ticks acquire Babesia infection from an infected animal and then pass the agent to their offspring through the ovaries. The protozoan is passed to susceptible cattle by adult ticks and nymphs. Most infections result in intravascular and extravascular hemolysis and in kidney and liver failure. See Chapter 37 for more detail.
A small proportion of Babesia infections cause acute encephalitis.2,3 CNS signs begin suddenly and include fever (41.7° C [107° F]), anorexia, depression, ataxia, conscious proprioceptive deficits, mania, convulsions, and coma. Sudden death is occasionally observed. The nervous system signs are accompanied by engorgement of the scleral vessels, icterus, proteinuria, and hemoglobinuria. Encephalopathic diseases that closely resemble babesiosis include rabies, nervous coccidiosis, polioencephalomalacia, lead poisoning, bovine herpesvirus encephalitis, theileriasis, heartwater disease, salt poisoning, and chlorinated hydrocarbon toxicity.
The pathogenesis of the CNS signs is unclear; however, possible causes include capillary thrombosis and infarction, disseminated intravascular coagulation, anoxic encephalopathy, and direct invasion of the CNS by the parasite. Thrombi are disseminated throughout the CNS. Expression of parasite proteins on the surface of infected RBCs facilitates binding of RBCs to capillary endothelial cells.4 The proclivity of Babesia-infected RBCs for binding to brain capillaries, particularly those in the cerebellum, is supported by the high incidence of parasite-positive RBCs found in the brains of infected cattle.5 These findings and the observation of increased prothrombin and partial thromboplastin times, thrombocytopenia, and decreased fibrinogen concentrations suggest that disseminated intravascular coagulation may play a role in the pathogenesis of the CNS disease.6-8 Vascular blockage in the CNS, caused by severe sludging of RBCs within brain capillaries, appears to be central to the pathogenesis of neurologic disease in infected animals.9 Studies of Babesia ovis in sheep, in which neurologic disease did not occur, failed to demonstrate RBC blockage of brain capillaries.10 This further supports the belief that CNS vascular pathology is key to the development of neurologic signs in bovine babesiosis.
Babesia encephalitis is a reportable disease in the United States. Suspected cases should be referred to the appropriate state and federal authorities.
Ehrlichia (Cowdria, Rickettsia) ruminantium
Infection (Heartwater Disease)
■ Definition, Etiology, and Epidemiology Ebrlicbia ruminantium is a rickettsial parasite that causes a fatal encephalitis in goats, sheep, and cattle.1 The disease originated in sub-Saharan Africa and has spread to cattle in the West Indies (Guadeloupe, Antigua, and Marie Galante).2 E. ruminantium is transmitted by Amblyomma ticks.3,4 Although a number of Amblyomma species have been implicated in the transmission of heartwater disease, the ones that cause most cases are Ambly- omma variegatum and Amblyomma bebraeum. The intermittent feeding behavior of the tick makes it particularly difficult to control. Amblyomma ticks require three separate blood meals to complete their life cycle. The gravid females fall from the host and lay the eggs in rotting vegetation, particularly in areas where the hosts are bedded for the evening. Recently hatched larvae crawl onto foliage and await a host. After their first feed the larvae detach, molt into nymphs, and await a second host. After refeeding, the nymphs detach and molt into adults. The adults remain under rotting vegetation until they are activated by carbon dioxide exhaled by a large mammal. They are further attracted to the host by pheromones from male ticks that remain permanently attached to the host. Animals that do not have male tick infestations are poor attractants for nongravid females. Once attached to the proper host, the females seek the male, breed, feed, and fall from the host when it lies down for the evening. Ticks that feed from Ebrlicbia-infected hosts develop ovarian infections and transmit the agent to their offspring. This serves to perpetuate the agent over successive seasons.5
Many species of vertebrates, including snakes, iguanas, lizards, and birds, are reservoirs for E. ruminantium because these animals may serve as the first two hosts for the Amblyomma tick.6 A vector-wildlife cycle facilitates the survival of E. ruminantium even when domestic livestock are absent from the environment.7
■ Pathogenesis After inoculation into a ruminant, the rickettsial agent infects reticuloendothelial cells and proliferates by binary fission within membrane-bound vacuoles.8,9 Release of the parasite from degenerating macrophages and neutrophils causes successive waves of parasitemia that infect endothelial cells and cause vasculitis.9,10 Nervous system lesions may be caused by permeability changes in the cerebral capillaries. Changes in the other soft tissues include hydropericardium, hydrothorax, and subcutaneous edema.1
Except for Angora goats, which are highly susceptible to Ebrlicbia infection, animals reared in indigenous areas usually have a high level of immunity and do not succumb to the infection.11 Animals that survive the initial infection no longer show symptoms but remain rickettsemic for as long as 223 days (sheep), 246 days (cattle), and 8 days (goats). Calves younger than 3 weeks of age, lambs younger than 8 days of age, and kids younger than 6 weeks of age are inherently resistant to E. ruminantium infection, regardless of the amount of colostral 1213
protection they have received.12,13
■ Clinical Signs Animals with the peracute form of E. ruminantium infection die suddenly without premonitory signs. The acute form of the disease is characterized initially by fever, anorexia, depression, and respiratory distress. Cyanosis may also be noted. Nervous system signs, which may appear within a few days, include hyperesthesia, snapping closure of the eyelids, rapid extension of the tongue, behavioral changes, muscular fasciculations, hypermetria, ataxia, conscious proprioceptive deficits, and head pressing. As the disease progresses, the animals become recumbent and comatose. Convulsions may occur in the terminal phase. These episodes are characterized by opisthotonos, nystagmus, chewing movements, and frothing at the mouth. Mild forms of the disease are characterized by transient diarrhea, malaise, and fever, with no CNS involvement. The mortality rate in sheep ranges from 6% to 80%. Animals that recover are immune to reinfection for at least 58 months.14 Losses may reach 60% of susceptible cattle and 40% of goats. The mortality rate among Angora goats may exceed 90%. Enterotoxemia caused by Clostridium perfringens type D is important in the differential diagnosis in small ruminants.
■ Pathology Pathologic changes of heartwater disease include hydropericardium, hydroperitoneum, hydrothorax, pulmonary edema, perirenal edema, hemorrhages in the pleura and peritoneum, and hemorrhagic enteritis. Microscopic changes include microgliosis, necrotizing vasculitis in the brain, hemorrhage, edema of the neuropil, microcavitation, and focal
necrosis of the granular layer of the cerebellum. In clinical cases, the parasite can be definitively diagnosed only through biopsy of the cerebral cortex or by collection of the cortical tissues at postmortem examination.15 Simple techniques for collection of such biopsy specimens have been described.16-18 Squash preparations of the biopsy sample should be stained with either methyl green pyronine or Giemsa stain before 19 20
microscopic examination of the tissues.,
Diagnostic tests developed to identify infected livestock include an IFAT in which infected bovine aortic endothelial cells serve as indicators, several ELISAs, and tests involving PCR methodology.21-24 Of these, PCR is the most accurate.
■ Treatment and Control The administration of oxytetracycline (6 to 10 mg/kg IV twice daily for 3 or 4 days) may be beneficial for treatment of the early stages of the disease. The long-acting formulation of oxytetracycline is also effective, but for best results it should be administered as soon as the animal becomes febrile. Treatment is usually futile if the first dose of oxytetracycline is administered after the onset of neurologic signs.25 Despite the depository nature of the long-acting formulation, two or more doses 48 hours apart are necessary to achieve a good clinical response. Cattle should be re-treated if they develop a fever after the first dose has been administered.26 Many animals with CNS signs die despite intensive antibiotic therapy. Angora goats are highly susceptible to heartwater disease. In South Africa, Angora producers routinely treat all animals every 14 days during the summer with oxytetracycline. Animals that remain essentially tick free never develop adequate immunity to the Ehrlichia organism.
A small degree of tick infestation and exposure of the animals to low numbers of the agent, combined with judicious oxytetracycline therapy, appears to favor the development of immunity over time.27 One method of immunization is to use a controlled infection of a virulent strain (Onderstepoort Ball 3 strain) of the Ehrlichia organism and treatment with long-acting oxytetracycline (800 mg per adult goat) at the beginning of clinical disease and 10 days later.28 Vaccination with this isolate produces immunity against exposure to homologous, but not heterologous, strains of Ehrlichiar'-'' More recent strategies have included vaccination with inactivated organisms, live attenuated organisms, and fragments of E. ruminantium nucleic acid, but none has produced great success.32-36
Control of ticks on pastures is the most desirable means of controlling heartwater disease. Complete eradication of the ticks in most regions of sub-Saharan Africa is neither possible nor desirable. Cattle that are reared in areas where Ehrlichia infection is endemic acquire immunity over time. In these areas, the ticks should be sufficiently abundant to enable a low level of heartwater infections in most cattle but not so populous as to introduce severe, overwhelming infections. Integrated methods for tick control have been recommended. These include exclusion of wildlife from paddocks, artificial induction of host resistance to ticks, application of insecticidal ear tags, and conventional acaricide application. Application of insecticides as the sole method of tick control has not proved highly effective. Resistance to the acaricides may develop with prolonged use.
Heartwater is a reportable disease in the United States. Suspected cases should be referred to the appropriate state and federal authorities.
Cerebral Theileriasis (Turning Sickness; Draaisiekte; East Coast Fever; Corridor Disease; January Disease; Tropical Fever)
■ Definition, Etiology, and Epidemiology Cerebral theileriasis is an encephalitic disease of cattle caused by the piroplasma parasites Theileria annulata and Theileria parva.1,1 Theileriasis occurs mainly in Africa (Kenya and Tanganyika) and India, where it is characterized by a high mortality rate and is known as East Coast Fever. Theileria sergenti is found in Korea andJapan. Theileria mutans, a parasite of cattle in the southwestern United States, is relatively nonpathogenic.3 A mild form of theileriasis, locally called “January disease,” occurs in cattle in Mozambique. This condition is caused by the subspecies T. parva bovis. Corridor disease is caused by T. parva lawrencei, and T. annulata is the cause of Mediterranean Coast fever or tropical theileriasis.
European breeds of cattle (Bos taurus) develop a more severe disease than do comparably infected Indian breeds (Bos indicus). Both types of cattle appear to be equally susceptible to infection but differ in their ability to control the course of disease.4 Theileria organisms are transmitted to susceptible cattle by the ticks Hyalomma anatolicum, Hyalomma lusitanicum, and A. hebraeum.5
■ Pathogenesis Trypanosoma spp. manipulate the immune system both to evade destruction and to promulgate infection. Organisms escape the host immune response by altering immune system activation so that infected cells avoid recognition and destruction.6-8 Clonal expansion of infected lymphocytes is promoted, and these cells disseminate throughout the body.9
■ Clinical Signs The clinical signs of Theileria infections include lymphadenopathy, nasal discharge, lacrimation, tachycardia, fever, subcutaneous edema of the face, gangrenous dermatitis, sloughing of facial skin, dyspnea, pallor, CNS disease, and emaciation.10 The neurologic syndrome is characterized by ataxia, hypermetria, conscious proprioceptive deficits, depression, head pressing, hyperesthesia, blindness, nystagmus, circling, and aggressiveness. In the terminal stage, the animals become recumbent and develop opisthotonos, tonic-clonic seizures, and coma. In rare cases the parasite may localize in the spinal cord. The CNS signs result from vasculitis and lymphocytic inflammation of the brain.11-13 Clinically recovered animals remain persistently infected.
■ Pathology The postmortem lesions of theileriasis include capillary engorgement, scattered punctate hemorrhages on the surface of the brain, thrombosis of the meningeal vessels, hemorrhage in the cerebral ventricles, pulmonary edema, peripheral lymphadenopathy, and infarctions of the kidney and spleen. The brain of an affected animals appears to yel- lowish.13 On microscopy, blue cytoplasmic inclusion bodies (Koch blue bodies) are observed in the lymphocytes adjacent to the hemorrhagic areas.
The nervous form of theileriasis is difficult to diagnose definitively because the parasite is only sporadically visible in sections of nervous tissue from the infected animals. Parasitemia is occasionally detected in microscopic examination of blood smears from infected calves. Reliable blood tests are not currently available. Biopsy of the cerebral cortex has been recommended as a confirmatory test for the disease. The CSF of affected animals is normal or has an increased protein concentration ranging from 1400 to 12,452 mg/dL, with normal numbers of WBCs.12
■ Treatment and Control Parvaquone (10 to 20 mg/kg IM in two injections 48 hours apart) and buparvaquone (at a single dose of 2.5 mg/kg) are approximately 90% effective for the treatment of theileriasis.14 Some studies have shown buparvaquone to be the most effective treatment.15,16 Parvaquone can be combined with furosemide, which improves survival of cattle that develop pulmonary edema, a common consequence of theileriasis.17 Menoctone (10 mg/kg IV or IM) is also curative. Single doses were effective in one study, but repeating the treatment daily for 5 days eliminated pOsttherapeutic recrudescence.18
Prevention of Theileria infection has been based largely on the use of live attenuated vaccines, which produce a solid, cell- mediated immunity.19 These vaccines are used in an “infection and treatment” manner, whereby vaccination must be combined with administration of long-acting tetracyclines.20 Recent efforts at vaccine development have focused on the creation of subunit vaccines involving a variety of Theileria antigens.
The naphthoquinones are the preferred chemotherapeutic agents in vaccinated animals and include parvaquone, halo- funginone lactate, and buparvaquone.
Tick control is vital for prevention. Weekly spraying with coumaphos and insertion of ear tags impregnated with cyper- methrin have been effective for controlling ticks and preventing Theileria infections in calves in Tanzania. If the tags are removed, the calves become infected with T. parva within 3 months. Woodford21 suggested that intensive tick control could minimize the incidence of theileriasis until the calves can be successfully vaccinated. However, inadequate or incorrect application of products and tick resistance to chemicals present challenges to effective tick control. The disease is exotic to North America.
Cerebral Trypanosomiasis (Sleeping Sickness) alterations included an increase in total protein (range, 37 to 44mg/dL) and pleocytosis (range, 0 to 3060 mononuclear cells per microliter). The abnormalities in the CSF may be present in infected cattle not currently displaying clinical neurologic signs. Antitrypanosomal antibody may be found in the CSF of affected cattle with the use of IFAT.
■ Pathology The pathologic lesions of trypanosomiasis are a nonsuppurative encephalomyelitis, serosanguinous pericardial fluid, serosal hemorrhages, pulmonary edema, centrilobular coagulative necrosis, splenomegaly, necrotizing myocarditis, and glomerulonephritis. Macroscopic lesions of the CNS include subtle thickening and grayish discoloration of the meninges. The meningeal vessels are congested. Microscopic lesions of the CNS reflect moderate diffuse meningoencephalitis. The pathogenesis of the encephalitis is not understood.
Trypanosomes may be cultured from the blood and CSF of infected cattle. The number of hematogenous parasites is highest in animals with a dual infection by T. congolense and T brucei. The clinical diagnosis of trypanosomiasis may be confirmed by inoculating blood or CSF specimens into laboratory mice and observing the recipients for parasitemia with direct darkfield examination of the patient's blood. Identification of motile trypanosomes in the buffy coat zone of a microhematocrit capillary tube with darkfield illumination is apparently the most accurate of all diagnostic methods. Trypanosomes can be differentiated by their morphologic features, manner of attachment to erythrocytes, and type of motility. Infected cattle develop acquired resistance to homologous, but not heterologous, isolates of trypanosomes.
■ Treatment and Control Of the drugs available for the treatment of trypanosomiasis, isometamidium chloride is most often used. The recommended dose ranges from 0.25 to 1 mg/ kg IM; a single dose of 1 mg/kg exerts a protective effect for up to 6 months. The higher dose (1 mg/kg) is necessary to increase weight gain and prevent recurrent infection. The treatment was particularly effective when combined with weekly surveillance, followed by treatment of animals confirmed to be infected. Side effects of isometamidium include tachycardia, salivation, lacrimation, pollakiuria, muscle fasciculations, convulsions, diarrhea, and, in rare cases, death. The drug apparently does not cause abortion in pregnant cows or otherwise affect the calf.
Other drugs used include suramin sodium, diminazene aceturate (7 mg/kg), quinapyramine sulfate, and homidium chloride (1 mg/kg). These drugs provide residual protection from reinfection for approximately 2 months; however, recurrent infection and resistance, especially to diminazene, have been reported.11-14 Combinations of these drugs (e.g., diminazene followed by isometamidium) reduce the number of resistance- related therapeutic failures. Relapses also occur, undoubtedly because many of the chemoprophylactic drugs are unable to penetrate the blood-brain barrier in sufficient concentrations to eliminate the parasite in the CNS tissues.
Because of the variable responses to treatment and the ability of Trypanosoma organisms to develop drug resistance, all control programs must include methods of controlling the tsetse fly. Because the tsetse fly prefers to feed from the ventral torso or the legs, insecticide-impregnated ear tags have been ineffective for preventing infection; other means of application of insecticide are necessary. Other fly control methods include aerial spraying with endosulfan, ground-based spraying with 4% dichlorodiphenyltrichloroethane (DDT), or use of scented insecticidal traps. However, cost and environmental concerns have limited the usefulness of these techniques.
Selection of resistant lines of cattle is possible. The taurine N'Dama and West African shorthorn breeds have innate resistance to trypanosomiasis and are the sole breeds of cattle in areas of tsetse fly range.16 Resistant breeds of small ruminants include the Djallonke, Red Maasai, Blackhead Persian, and East African sheep and goats.17 Imported breeds of livestock usually cannot be maintained, even in areas of low tsetse fly risk, without intensive drug therapy.18