The Retinoblastoma and p53 Proteins Are the Main "Gatekeepers" for the Cell Cycle
Retinoblastoma is a rare, hereditary, childhood cancer of the retina of the eye. Despite its rarity and that it cannot be induced in mice, retinoblastoma has played an important role in the study of cancer.
A statistical study of the disease in the early 1970s provided the best evidence then available that human cancer is a genetic disease. Alfred Knudsen showed that children with retinoblastoma typically inherit one mutant copy from a parent (a “germ line mutation”), but then required a second somatic mutation in cells giving rise to the retina. Knudsen’s “two-hit hypothesis” was a forerunner to the idea that cancer develops by the accumulation of mutations in a cell lineage. (Retinoblastoma tumors do require the accumulation ofadditional mutations beyond the two retinoblastoma genes being mutant.) Subsequently, the retinoblastoma gene, RB, was the first tumor suppressor gene to be cloned. Study of the encoded protein, pRB, showed that it played a central role in controlling the transition from Gl to S phase of the cell cycle.The RB protein is a repressor of a transcription factor whose activity is required for the cell to enter S phase from Gl (Figure 2-8). The transcription factor is E2F, which controls the expression of a wide variety of genes/proteins required for DNA synthesis, including cyclin A, CDKl (see Figure 2-4),
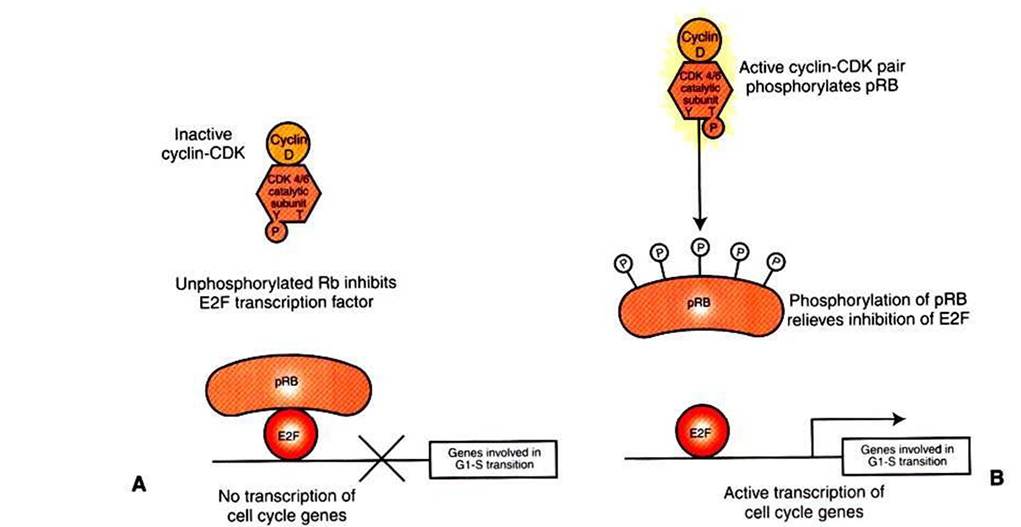
FIGURE 2-8 Retinoblastoma protein and the GVto-S transition. A, In quiescent cells or cells early in G1, retinoblastoma protein (pRB) exists in a nonphosphorylated state that is a direct inhibitor of the E2F transcription factor.The principal CDK pair of G1, cyclin D with CDK4 or CDK6, phosphorylates pRB, releasing its inhibition of E2F. Bf Activated E2F then participates in the expression of a variety of genes required for S phase, including the cyclins and CDKs of S phase and subunits of DNA polymerase.
and subunits of DNA polymerase. The RB protein is a potent inhibitor of E2F only when it is bound to E2F directly, which requires the RB protein to be in an unphosphorylated state (see Figures 1-1, B, and 1-17). The repression of E2F is released by phosphorylation of RB by cyclin-CDK pairs operating early in Gl in the cell cycle. As discussed, growth factor stimulation of the MAP kinase pathway leads to expression of cyclin D (see Figure 2-5), which in turn makes a pair with either CDK4 or CDK6 to make an active CDK. One of the substrates for cyclin D∕CDK4,6 is the RB protein. When RB is phosphorylated by CDK4,6, it releases from E2F, allowing this transcription factor to promote RNA polymerase activity on genes with E2F promoter regions (Figure 2-8). It is this release of inhibition by CDK-mediated phosphorylation of RB that constitutes the molecular mechanism underlying the R-point “decision" to divide late in Gl mentioned earlier and shown in Figure 2-2. Ifboth copies of RB are mutant, as in retinoblastoma, there will be no active repressor molecules to bind to E2F, and the decision will always be to divide, regardless of other conditions. E2F then promotes uncontrolled expression of S-phase genes whether or not CDK4,6 has been activated (in part) by growth factors and adhesion, thus making a contribution to unregulated growth and to cancer. Conversely, in its normal, nonmutant form, RB tends to suppress tumor formation by acting as a gatekeeper, only allowing the cell “to cross the border" between Gl and into S phase if normal growth factor and adhesion signals are received. Thus, pRB plays a crucial gatekeeper role in healthy, normal cell cycle control.
The other crucial gatekeeper between Gl and S phase is p53. Unlike pRB, p53 does not participate in healthy cell cycles; p53 is only active in response to cell damage, usually DNA damage, or stress, such as low O2 concentration or oncogene activation (Figure 2-9). The role of p53 is to ensure that stressed/damaged cells are either repaired or, if not, commit suicide before being allowed to replicate their DNA.
As a gatekeeper, p53s mechanism is also more direct than pRB; p53 is itself a transcription factor, and p53 activation stimulates the expression of a protein that is a powerful general inhibitor of all the cydin/CDK engines. As a transcription factor, p53 also mediates the expression of genes that encode stimulators of cell death, as discussed shortly. Whether the cell responds to p53 by cell cycle arrest to allow repair, or by committing suicide, depends on multiple factors, but presence of an oncogene is among the most important. Normally, the cell cycle arrest activity of p53 is dominant to its death-inducing activity. However, in the presence of oncogenes, including ιιιyct suicide is favored. This illustrates clearly the normal tumor suppressor activity of p53: although a cell expressing an oncogene will tend toward increased proliferation, the same oncogene, acting through p53, activates a death pathway to prevent expansion of the mutant cell population.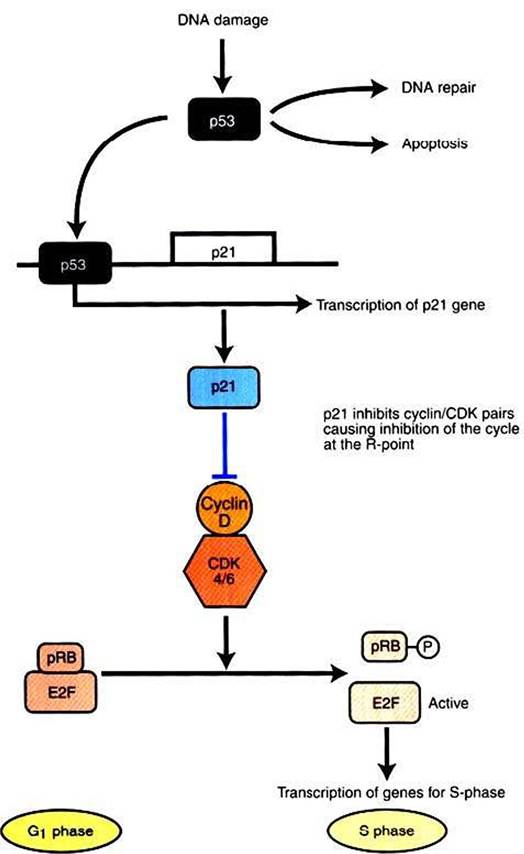
FIGURE 2-9 p53 and the response to DNA damage.
Normally, p53 is maintained at low levels in the cell by continuous synthesis and breakdown. DNA damage inhibits breakdown, allowing p53 to build up to functional levels. p53 is itself a transcription factor, and its targets include p21, a potent inhibitor of all cyclin-CDK pairs.Thus, upregulation of p53 brings the cell cycle to a halt, typically by inhibiting phosphorylation of pRb, as shown here. Subsequently, if the DNA is repaired, p53 returns to low concentration. If the DNA remains damaged, p53 leads to an apoptotic response by mediating expression of pro-apoptotic proteins, as described in the text.
The activation of p53 occurs in part through mechanisms familiar from previous examples of protein control, including phosphorylation and binding with other proteins. In addition, p53 activity is also regulated simply by an increase in its concentration within the cell.
That is, p53 is normally synthesized at a steady but slow rate throughout the cell cycle and is normally degraded at a similar rate. In healthy cells the halflife for a p53 molecule is about 30 minutes, but this increases threefold to sevenfold in response to DNA damage. Even one double-strand break in DNA has been shown to increase p53 concentration rapidly in some cells. Again, it is clear how p53 serves as both a gatekeeper and a tumor suppressor. Activated p53 prevents a cell with DNA damage from crossing the Gl-S boundary (its gatekeeper function), which in turn prevents mutant cells from being allowed to accumulate additional mutations (its tumor suppressor function).However, if the p53 gene suffers a loss-of-function mutation and the protein cannot act as a transcription factor, a damaged cell will indeed be able to divide, increasing the probability of accumulating further damage and leading to possible cancer. Thus, ρ53 is one of the most important single genes and proteins involved in human cancers; in 1993 the journal Science even named it “Molecule of the Year.” About 50% of human tumors have a mutation in ρ53, with most of these eliminating DNA binding, disabling its transcription factor activity. When the ρ53 gene was “knocked out” in mice, 74% of the animals developed cancers by 6 months of age (young adult). Among experimental mice that had one or two normal copies of the gene, only 1 in 100 animals developed a tumor by 9 months.
In addition to a checkpoint for S phase in which DNA damage provides an important regulatory signal, the other major checkpoint occurs during mitosis. This checkpoint responds to mitotic spindle abnormalities or damage and to abnormalities in the array of chromosomes within the spindle. Here again, one can easily see how mutations that disrupted such “safety interlocks” could lead to further damage, by segregating both replicated chromosomes into one daughter cell, for example, with no copy of that chromosome in the other daughter cell. This would lead directly to aneuploidy. Among human cancers, colon cancer is frequently found to have mutations in mitotic checkpoint genes.
However, we leave the topic of mitotic checkpoints at this somewhat intuitive level and do not address the molecular mechanisms in detail. Such an effort would require a lengthy background discussion of the structure, functions, and control of the microtubule-based mitotic spindle, more suitable for a course in cell biology than animal physiology. Instead, we now discuss the controls on cell growth other than proliferation and briefly summarize what is known about programmed cell death and the control of cell life span.