Adrenal Glands
Kelsey A. Hart
Physiology of the Adrenal Gland and Hypothalamic-Pituitary-Adrenal Axis
As in other species, in the horse the paired adrenal glands lay immediately Craniomedial to the kidneys.
Equine adrenal glands weigh approximately 15 g each and are approximately 8 to 10 cm in length, 3 to 4 cm wide, and 1.5 cm thick, though there is much variation in adrenal size and shape among individual horses. Each gland is divided into an outer cortex that secretes corticosteroids and an inner medulla that secretes catecholamines.The adrenal medulla functions as a neuroendocrine organ and is essentially a modified sympathetic ganglion. Preganglionic sympathetic neurons extend from the central nervous system and synapse directly on adrenal medullary chromaffin cells; when stimulated, these cells release catecholamines directly in the systemic circulation to produce a global sympathetic response. In the horse, the primary adrenal medullary catecholamine appears to be epinephrine, though norepinephrine and dopamine are also produced.1 Diseases affecting the adrenal medulla are rare in horses—as in all species—though adrenal medullary neoplasms are reported.
The adrenal cortices are divided into three cellular zones: (1) the outer zona glomerulosa; (2) the middle zona fasiculata, which is the largest zone and comprises approximately 75% of the weight of the gland; and (3) the narrow inner zona reticularis. Cells in the zona glomerulosa are primarily responsible for the secretion of mineralocorticoids (e.g., aldosterone) in response to hypotension and electrolyte derangements (hyperkalemia, hyponatremia). Zona fasiculata cells synthesize and secrete glucocorticoids (e.g., cortisol, corticosterone, 11-deoxycortisol) in response to stimulation by adrenocorticotropic hormone (ACTH) after activation of the hypothalamic-pituitary-adrenal (HPA) axis (Fig.
41.8) by physiologic or pathophysiologic stressors. The predominant circulating glucocorticoid in the horse is cortisol.2 Cells in the zona reticularis also secrete small amounts of glucocorticoids but primarily produce adrenal androgens such as dehydroepiandrosterone (DHEA) and androstenedione. All corticosteroid hormones are structurally similar with a common sterol backbone and one 5-carbon and three 6-carbon rings, and they share a common synthetic pathway originating from cholesterol (Fig. 41.9). The specific corticosteroids produced by a particular adrenocortical cell depend on which specific biosynthetic enzymes are expressed in that cell (see Fig. 41.9), which varies between the three functional zones.All steroid hormones are lipophilic and thus are transported in the plasma predominantly bound to plasma proteins, including albumin and steroid-binding globulins such as cortisol- binding globulin (CBG). However, as steroid hormone receptors are located in the cytoplasm of steroid-responsive cells, it is only the free, unbound portion of circulating steroid hormones that is available to enter cells via diffusion across the plasma membrane to bind these intracellular receptors. Binding of the steroid hormone to the receptor causes conformational changes that allow dissociation of regulatory heat shock proteins, permitting the hormone-receptor complex to localize to the nucleus, bind DNA at hormone-response-elements (HREs), and upregulate or downregulate transcription of steroid hormone-responsive genes.
Glucocorticoids are the most abundant circulating adrenal corticosteroid hormone and play an integral role in the endocrine response to stress. The HPA axis (see Fig. 41.8) is activated when physiologic, pathophysiologic, or environmental stressors activate peripheral and central nervous system components, whose signals are then interpreted and integrated in the hypothalamus. This stimulates hypothalamic paraventricular nuclei, resulting in the release of corticotropin-releasing hormone (CRH) into the hypothalamic-hypophyseal portal vessels.
CRH then acts locally in the adjacent anterior pituitary gland to stimulate CRH receptors on the cell surface of pituitary corticotroph cells, resulting in the release of adrenocorticotropic hormone (ACTH, corticotrophin) into the systemic circulation.ACTH binds cell surface receptors (melanocortin 2 receptor, MC2R) on adrenocortical cells and stimulates the adrenal glands to synthesize and secrete cortisol. MC2R is a G protein-coupled transmembrane receptor that acts via adenylate cyclase to increase cyclic AMP levels, which then activate biosynthetic enzymes necessary for cortisol synthesis, including 3-β-hydroxysteroid dehydrogenase (3-β-HSD), 17-α-hydroxylase, 21-α-hydroxylase, and 11-β-hydroxylase (Fig. 41.9). This latter enzyme catalyzes the final step in cortisol synthesis from 11-deoxycortisol and is present only in glucocorticoid-producing cells.3
Cortisol is not stored in adrenocortical cells but rather is secreted into the systemic circulation immediately following ACTH-induced synthesis. In most adult mammals, including
FIG. 41.8 Hypothalamic-pituitary-adrenal (HPA) axis.
Stimulatory interactions are illustrated with solid arrows and + signs, and inhibitory interactions (negative feedback) are illustrated with dashed arrows and — signs. ACTH, Adrenocorticotropic hormone; CRH, corticotropic releasing hormone. (From Hart KA, Barton MH: Adrenocortical insufficiency in horses and foals. Vet Clin North Am Equine Pract 27:19-34, 2011.)
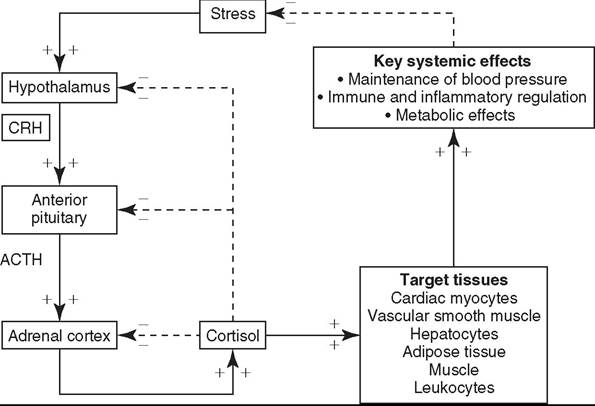
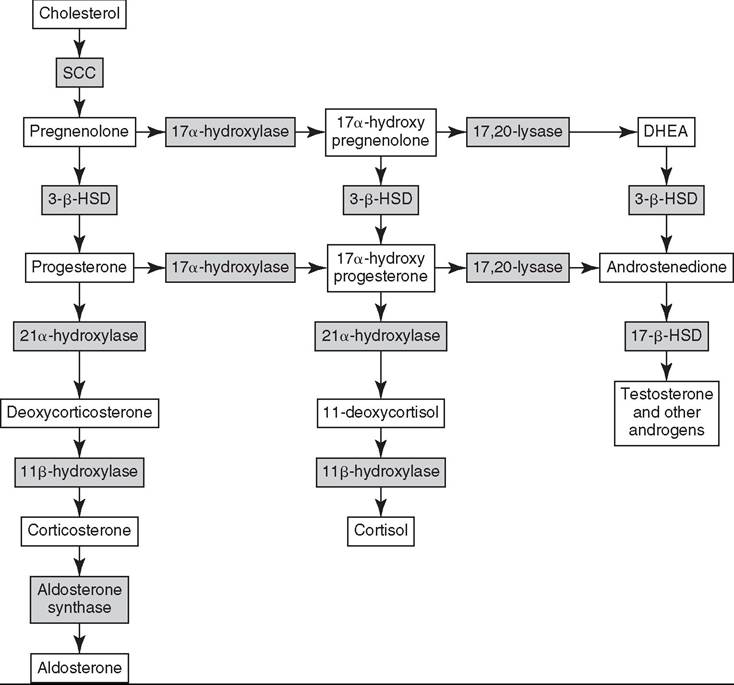
FIG. 41.9 Biosynthetic pathway for adrenal corticosteroids. The enzymes responsible for catalyzing each biotransformation are shown in the gray boxes over the arrows. SCC, Side chain cleavage enzyme; 3-β-HSD, 3-β-hydroxysteroid dehydrogenase; DHEA, dehydroepiandrosterone. (From Hart KA, Barton MH.
2011. Adrenocortical insufficiency in horses and foals. Vet Clin North Am Equine Pract. 27:19-34)horses, approximately 90% to 95% of circulating cortisol is bound to CBG and albumin.4-8 Many cell types are sensitive to glucocorticoids, permitting cortisol to exert diverse effects necessary for maintenance of homeostasis and stress responses in both health and disease. Essential glucocorticoid-mediated physiologic responses include maintenance of blood pressure, provision of energy to tissues, and control of an appropriate inflammatory response. The sum of cortisol’s systemic effects serves to reduce the physiologic stress that initially activated the HPA axis, to ultimately restore basal HPA axis tone. In addition, cortisol itself acts via negative feedback mechanisms at hypothalamic, pituitary, and adrenal levels to further downregu- late HPA axis activity. Thus, with an intact HPA axis, plasma cortisol concentrations are maintained at a level appropriate for the existing degree of physiologic stress (see Fig. 41.8).
Cortisol secretion patterns in adult horses exhibit both ultradian (pulsatile) and circadian rhythms with peak secretion in the morning and the nadir in the evening, similar to many other species.9-12 These ultradian and circadian rhythms are easily disrupted in horses by simple routine changes and illness.9,12 Cortisol responses to ACTH stimulation testing in adult horses are described and suggest adult horses show a comparable dose-dependent effect of exogenous ACTH on cortisol concentrations as is described in other species.11,1315 In general, an approximately twofold to fivefold increase in cortisol concentration is observed within 30 to 90 minutes after IV administration of a 0.1 to 10 μg∕kg dose of exogenous ACTH (cosyntropin) in healthy adult horses13,15; the magnitude of cortisol increase varies with the ACTH dose administered.
HPA axis function in the foal differs from both adult horses and other species in several key ways.
Compelling evidence shows that maturation of the HPA axis occurs in the days just before parturition and continues during the first several weeks of life, much later than is described in other species.16-19 Premature foals have lower serum cortisol and higher ACTH concentrations than full-term foals immediately postpartum.19 These low baseline cortisol and concurrent high ACTH concentrations in premature foals imply that foals may have impaired adrenocortical sensitivity to ACTH, limited cortisol synthetic capacity, or both, as compared with adult horses.Data from ACTH stimulation test responses in foals support this theory, as premature foals show a blunted cortisol response to exogenous ACTH, with only a 28% increase in plasma cortisol 30 to 60 minutes following stimulation, as compared with a 208% increase in normal-term foals.16 Some evidence suggests that the fetal foal’s adrenal gland may be incapable of synthesizing cortisol and some other steroid hormones until late in gestation. In contrast to most other species, fetal foals do not express substantial levels of the key steroidogenic enzymes cholesterol side chain cleavage enzyme, 17α-hydroxylase, and 3-β-HSD (see Fig. 41.9) until just before parturition in the foal,20,21 much later than is described in other species.22
Furthermore, adrenocortical function may not be fully mature at birth even in full-term foals. Though increased at birth due to the stress of parturition, by 12 to 24 hours of age, mean basal cortisol concentrations are onefold to twofold lower in healthy neonatal foals than reported mean concentrations in healthy adult horses despite comparable or higher concurrent ACTH concentrations in foals.23-27 This apparent decreased cortisol response to endogenous ACTH in the full-term neonatal foal is supported by further evidence of limited cortisol responses to exogenous ACTH. Insulin-induced cortisol responses in foals within 12 hours of birth are less than half of the responses achieved in 7- to 14-day-old foals,28 and healthy-term neonatal foals aged 1 to 7 days are reported to have approximately half the magnitude of cortisol response to ACTH stimulation testing than adult horses.13,24 This decreased adrenocortical responsiveness appears to persist during the first few months of life, as 12-week-old foals show significantly greater cortisol responses to a low-dose (0.1 μg∕kg) ACTH stimulation test than younger foals.29
Finally, foals differ from adult horses in their plasma cortisol binding capacity and cortisol secretion and metabolism patterns.
In foals younger than 1 week of age, 30% to 60% of circulating cortisol is free, in contrast to 5% to 10% in adult horses.30 This is likely due to decreased CBG concentrations in foals, as has been shown in infants.31,32 Free cortisol is preferentially excreted and metabolized over bound cortisol,33 so it is not surprising that increased cortisol clearance rates in foals as compared with adult horses have recently been described.12 Healthy full-term foals appear to be able to increase their daily cortisol production rate to compensate for this increased clearance, though as serum total cortisol concentration at 1 week of age remains significantly lower in foals than adult horses,12 foals' capacity to further and appropriately increase cortisol production in severe stress may be limited. It is likely that impaired cortisol responses in neonatal foals result from multiple factors, including decreased cortisol synthetic capacity and increased cortisol clearance and possibly other as yet undefined difference in ACTH sensitivity or activity. Regardless of the cause, this evidence of fetal and neonatal HPA axis immaturity is certain to substantially impact foals' stress responses during the neonatal period.Mineralocorticoids (e.g., aldosterone) are produced in small quantities relative to glucocorticoids but have equally important physiologic roles. Angiotensin II—via activation of the renin- angiotensin-aldosterone system by hypovolemia/hypotension or increases in plasma osmolality—and increased plasma potassium concentration are the primary stimulants of aldosterone synthesis and secretion. ACTH is also necessary for aldosterone synthesis, but ACTH administration to healthy adult horses resulted in a decrease in plasma aldosterone concentration in contrast to increases seen in other species.34,35
Like glucocorticoids, aldosterone circulates in the plasma bound to albumin or steroid-binding globulins and primarily acts by binding cytosolic mineralocorticoid receptors to alter transcription of genes necessary for sodium and potassium transport. Mineralocorticoid receptors are predominantly expressed in sodium-transporting epithelia in the distal renal tubules and colon, with lesser expression in the rest of the intestinal tract and heart. Aldosterone induces transcription of an aldosterone-regulated kinase that increases activity of apical membrane sodium channels.36 The net effect of this is increased sodium flux across epithelial cells, resulting in increased renal and intestinal sodium and water resorption and concurrent stimulation of potassium excretion via the basolateral Na+/K+ ATP-ase.36 Mineralocorticoids are vital for appropriate fluid and electrolyte balance, and mineralocorticoid deficiency rapidly results in hyponatremia, hyperkalemia, and hypovolemia.
Reported resting and exercising plasma concentrations of aldosterone in horses have been described.37,38 In contrast to their limited cortisol responses to appropriate stimuli, newborn foals appear to be able to mount a substantial aldosterone response to hypovolemia/hyponatremia.19,39 In fact, one study suggested that the aldosterone response in foals appears to be exaggerated in comparison with that in adult horses and may reflect differences in tubular sensitivity to aldosterone between neonates and adults.39 However, well-established reference ranges for plasma aldosterone concentrations in foals are not currently available.
In adult males, circulating androgens are primarily of testicular origin, but in females more than half of plasma androgens may originate from the adrenal cortices. ACTH is necessary for the synthesis and secretion of adrenal androgens, but another factor (or factors) that is as yet unidentified is also required for adrenal androgen synthesis.11 Adrenal androgens are also transported in the plasma bound to albumin and CBG and act via cytosolic steroid hormone receptors to modulate gene transcription. DHEA and androstenedione can be extra- glandularly metabolized to active testosterone and estrogens and play a particular important role in early pubertal sexual development in most species. Androstenedione concentrations in adult horses are reported, and sex and age variations in adrenal androgen concentrations may be relevant but at present are poorly understood in horses and foals.26,40 Increased adrenal androgen production is postulated to play a role in mares with estrus-related behavior problems, though this has not been definitively proven.26
Adrenal Gland Disorders
Critical Illness-Related Corticosteroid Insufficiency
Critical illness-related corticosteroid insufficiency (CIRCI) (also known as relative adrenal insufficiency) has been described in septic foals and adult horses with colic. The condition is best understood in people and is defined as an insufficient cortisol response or inadequate cortisol activity for the existing degree of critical illness.41-45 CIRCI is discussed in detail in the later section on endocrine dysfunction in critical illness.
Non-CIRCI Adrenocortical Insufficiency in Adult Horses
Classic adrenocortical insufficiency characterized by glucocorticoid and mineralocorticoid insufficiency and unrelated to critical illness is not well defined in horses or foals, and Addison's disease—permanent adrenocortical insufficiency secondary to immune-mediated adrenocortical destruction—has not been reported. Transient adrenal insufficiency characterized by low basal ACTH and cortisol concentrations and impaired cortisol responses to ACTH stimulation testing has been described in one horse following abrupt cessation of long-term anabolic steroid supplementation.46 A syndrome of “adrenal exhaustion” resulting in lethargy, anorexia, and poor performance is also anecdotally described in racehorses and has been attributed to adrenal insufficiency associated with long-term anabolic steroid administration or chronic stress. Horses with presumptive adrenal exhaustion are often treated with exogenous ACTH for presumed adrenocortical stimulation, though measurement of basal cortisol concentrations and ACTH stimulation testing in race and endurance horses has not provided convincing evidence for adrenal insufficiency in these equine athletes.47,48 The potential for iatrogenic adrenal insufficiency associated with HPA axis suppression by exogenous steroid administration, though, should be considered in horses on chronic glucocorticoid or anabolic steroid supplementation, and care should be taken to avoid abrupt cessation of such therapy. Decreased cortisol responses to ACTH are also described in mares with abnormal estrus-related behavior,26 but the clinical significance of this behavior is not known.
In addition, the adrenal glands appear to be a shock organ in the horse and can also undergo hypoxic or ischemic damage during bouts of endotoxemia, sepsis, and systemic inflammatory response syndrome (SIRS).15,49 Thus, there is the theoretical potential for protracted or permanent adrenocortical dysfunction in animals who survive critical illness, though this specific manifestation is not reported in horses or foals to date. Adrenocortical insufficiency should be considered in horses with persistent lethargy, weight loss, hyponatremia, hypochloremia, hyperkalemia, or hypoglycemia, especially if they have recently received exogenous corticosteroids or recovered from critical illness. Failure to increase serum cortisol concentration greater than or equal to 1.5- to 2-fold baseline values by 30 minutes after a low-dose ACTH stimulation test (≤0.5 μg7kg IV) or greater than or equal to 3- to 4-fold baseline values by 90 to 120 minutes after high-dose ACTH stimulation test (≥1 μg'7lto RTA in horses.