Glomerular Filtration Rate Is Regulated by Systemic and Intrinsic Factors
The kidney normally maintains the GFR at a relatively constant level despite changes in systemic blood pressure and renal blood flow. The GFR is maintained within the physiological range by renal modulation of systemic blood pressure and intravascular volume and by intrinsic control of renal blood flow, glomerular capillary pressure, and K1.
Renal effects on systemic blood pressure and volume are mediated primarily through humoral factors, particularly the renin-angiotensin- aldosterone system. Intrinsic control of glomerular capillary perfusion is mediated by two autoregulatory systems that control the resistance to flow in the afferent and efferent arterioles: the myogenic reflex and Iubuloglomerular feedback.The renin-angiotensin-aldosterone system is an important regulator of GFR and renal blood flow. Renin is a hormone produced by specialized cells of the wall of the afferent arteriole, the granular extraglomerular mesangial cells, which are specialized juxtaglomerular cells. Renin release is stimulated by a decrease in renal perfusion pressure, most often caused by systemic hypotension. Renin catalyzes the transformation of angiotensinogen produced by the liver to angiotensin I. Angiotensin I is converted to the more active angiotensin II by angiotensin-converting enzyme (ACE), which is located primarily in the vascular endothelium of the lung. ACE is also present in the vascular endothelium of the kidneys and other organs, and thus the conversion of angiotensin I to angiotensin II may occur in extrapulmonary sites as well.
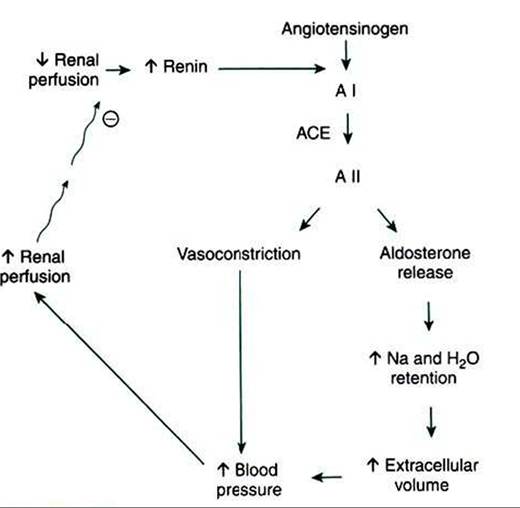
FIGURE 41-7 Schematic illustration of renin-angiotensin- aldosterone system. Circled minus sign represents inhibition. A I, Angiotensin I; ACE, angiotensin-converting enzyme; A II, angiotensin II.
Angiotensin II is a potent vasoconstrictor and thus directly increases systemic blood pressure and renal perfusion pressure. Angiotensin Il directly activates sodium uptake in both the proximal tubule and the collecting duct, and it stimulates the release of aldosterone from the adrenal gland and wιso- pressin from the pituitary. Thus, angiotensin II directly and indirectly enhances salt and water retention, intravascular volume increase, and vascular resistance, all of which contribute to increased systemic blood pressure and renal perfusion pressure. Renin release is suppressed by both the improved renal perfusion and the elevated plasma angiotensin II, creating a negative-feedback system that maintains renal perfusion and GFR within the physiological range (Figure 41-7).
Angiotensin II also stimulates production and release of at least two vasodilative renal prostaglandins: prostaglandin E2 (PGE2) and prostaglandin I2 (prostacyclin). This response is an important moderator of the renin-angiotensin-aldosterone system. The intrarenal production of these vasodilators counteracts the vasoconstrictive effect of angiotensin Il on the intrarenal vasculature and helps to maintain renal vascular resistance at normal or near-normal levels. Without this protective effect, generalized vasoconstriction would result in reduced renal blood flow and GFR, despite an elevation of systemic blood pressure.
Within the kidney itself, there appears to be direct control of glomerular capillary perfusion by two systems previously mentioned: the myogenic reflex and tubuloglomerular feedback. A mechanism of autoregulation of renal blood flow and GFR was proposed after the observation that glomerular arterioles respond to changes in arteriolar wall tension. This response is called the myogenic reflex; the result is almost immediate afferent arteriolar constriction after an increase in arteriolar wall tension and arteriolar dilation after a decrease in arteriolar wall tension.
Arcuate and interlobular arteries respond similarly. Thus, arteriolar dilation and constriction respectively decrease and increase the resistance to blood flow in the afferent arteriole. These changes in vascular resistance contribute to maintenance of GFR and renal blood flow at a constant level, despite marked alterations in the blood pressure in the renal artery. This reflex is independent of renal innervation but may be influenced by chemical mediators, such as nitric oxide.The second intrinsic control mechanism is tubuloglomerular feedback. To understand this concept, it is important to review the anatomical arrangement of an individual nephron (see Figure 4l-l). Specifically, recall that the distal tubule is closely associated with the glomerulus of the same nephron. An anatomically distinct cluster of epithelial cells, known as the macula densa, is located in the distal portion of the thick ascending limb of the loop of Henle. The macula densa is situated between the afferent and efferent arterioles and adjacent to the extraglomerular mesangial region. These four structures together are known as the juxtaglomerular apparatus, and they are closely related functionally as well as anatomically.
An increase in sodium chloride concentration in the tubule fluid at the macula densa causes a signaling cascade in the macula densa cells. This leads to generation of paracrine factors, such as nitric oxide, adenosine, and adenosine triphosphate (ATP), that suppress renin release from juxtaglomerular cells, increase resistance in the afferent arteriole, decrease glomerular capillary perfusion pressure, trigger mesangial cell contraction, and reduce Kl. These responses lead to reduced GFR in the individual nephron (single-nephron GFR), which prevents tubule fluid flow rates that exceed the tubule’s transport capacity and thus prevents excessive fluid and solute loss.
In addition, it is now evident that the endothelium itself contributes to local control of renal vascular tone by producing potent vasoconstrictors and vasodilators.
Endothelium- derived constricting factors include the potent vasoconstrictor endothelin thromboxane A2 (a metabolite Ofarachidonic acid) and angiotensin II. Endothelium-derived relaxing factors include nitric oxide (NO), prostacyclin, and PGE2. Of these, NO was the first endothelium-derived vasodilating agent to be identified, and thus endothelium-derived relaxing factor is sometimes used as a synonym for nitric oxide. NO is now known to have important protective effects on the kidney. NO is produced in the kidney by oxidation of ι -arginine, catalyzed by isoforms of NO synthase. NO prevents renal damage by quenching reactive oxygen species, thus inhibiting intrarenal vasoconstriction, glomerular hypertension, mesangial cell proliferation, and mesangial matrix production,In addition to controls exerted by the kidney itself, systemic factors can contribute to changes in GFR. These include systemic control of blood volume and vessel tone. Many hormones regulate blood volume. Angiotensin II, aldosterone, and vasopressin (antidiuretic hormone) enhance water and solute reabsorption by the kidney and thus increase blood volume. Atriai natriuretic peptides, produced in the cardiac atria, cause both natriuresis (sodium wasting) and diuresis (water wasting) and thereby reduce blood volume.
Systemic factors that affect vessel tone also affect systemic blood pressure, renal perfusion, and ultrafiltration. Vasopressin and circulating catecholamines can cause systemic vasoconstriction and increase blood pressure. Beta-adrenergic stimulation can activate the renin-angiotensin system, and alpha-adrenergic stimulation can cause renal vasoconstriction, which can both reduce and redistribute renal blood flow. In addition to altering renal perfusion, vasoconstrictors can affect the other determinant of GFR, the ultrafiltration coefficient Kf. Vasoconstrictors may cause contraction of the mesangial cells within the glomerulus and thus reduce the area available for filtration.
Because Kf is the product of the area available for filtration and the hydraulic permeability, mesangial cell contraction in vivo would reduce Kf and thus reduce GFR.Insulin-Iike growth factor and high dietary protein levels can enhance GFR. Insulin-Iike growth factor increases GFR in kidneys of normal animals and after experimental renal ischemia. Chronically high dietary protein intake causes sustained increases in renal blood flow and GFR. Renal blood flow and GFR are transiently elevated after a single high- protein meal. These observations are clinically relevant in the management of chronic renal insufficiency and renal failure. Although it may seem desirable to increase GFR by any means in patients with chronic renal disease, in fact the increase in GFR resulting from some high-protein diets can cause more rapid progression of glomerular injury and renal failure in animals and in humans.
In birds the GFR is more variable than in mammals, but the regulatory mechanisms are not well understood. Birds, unlike mammals, exhibit intermittent filtration in reptiliantype glomeruli; this occurs during dehydration and decreases the GFR. This may result from the release of arginine vasotocin, the avian analogue of mammalian arginine vasopressin, which decreases GFR in birds by causing constriction of the afferent arteriole of reptilian-type nephrons. Although some authors report that a juxtaglomerular apparatus is present in avian kidneys, the macula densa is either absent or rudimentary, and Iubuloglomerular feedback has not yet been demonstrated.